









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El síndrome anti-GQ1b fue introducido por Odaka y su grupo en 2001 siendo el primero de los síndromes antigangliósidos definidos en términos inmunológicos(1)(2) En este trabajo analizamos la evolución histórica de los síndromes antigangliósidos hasta la actualidad, revisamos conceptos clásicamente aceptados y reforzamos la idea de que es necesaria una clasificación unificada basada en conceptos actualizados.
Material y método.
Se buscaron en Pubmed los siguientes términos en inglés: síndrome de Guillain-Barré y gangliósidos. Como filtros adicionales se aplicaron “revisiones” y “texto completo”. Se obtuvieron 1665 y 1407 resultados respectivamente. Por su mayor interés científico, se incluyeron 25 resultados para su análisis. Se añadieron 7 artículos encontrados a través de citaciones de artículos inicialmente seleccionados (artículos “hijos”). En conjunto, 31 artículos objeto de la presente revisión.
Resultados
1. Hitos clínicos
En 1859, Jean Baptiste Octave Landry de Thézillat, médico francés, describió diez casos de debilidad de curso agudo y patrón ascendente. Más tarde, en 1876, Karl Freiderich Otto Westphal acuñó formalmente el término de “parálisis aguda ascendente” en cuatro pacientes que fallecieron finalmente de insuficiencia respiratoria. En 1892, William Osler hizo una cuidadosa descripción del síndrome, reseñando que los pacientes que lo presentaban habían sufrido un cuadro febril y/o infeccioso previamente. Lo denominó "polineuritis febril aguda"(1) . Fue en 1916 cuando Georges Guillain, Jean-Alexandre Barré y André Strohl, publicaron los primeros casos de “parálisis aguda arrefléxica con incremento de proteínas en líquido cefalorraquídeo”, el clásico síndrome de Guillain-Barré (SGB) (3).
En 1951, Ewing Robert Bickerstaff y Philip Cyril Powter Cloake describieron la triada oftalmoplejía, ataxia y alteración del nivel de conciencia. Lo hicieron de forma independiente y sin vincular esta triada al SGB(4)(5)
Finalmente, en 1956, Charles Miller-Fisher describió la última de las triadas clásicas, oftalmoplejía, ataxia y arreflexia agudas, dándole nombre al síndrome que continúa ostentando hasta la actualidad(6) .
Sin embargo, Guillain se adelantó a sus colegas Bickerstaff y Fisher, siendo pionero en intuir que todos estos síndromes eran variantes de una misma entidad. En un simposio celebrado en Bélgica en 1938 describiría variantes como(3) “la forma inferior”, “la forma mixta espinal y mesencefálica” y “la forma mesencefálica pura”. Éstas últimas corresponderían al síndrome de Miller-Fisher y al síndrome de Bickerstaff respectivamente(1)(3) .
En 1981 Asbury y su grupo establecieron formalmente los primeros criterios clínicos diagnósticos para el SGB(6) Dichos criterios se revisaron y actualizaron en 2014 por el grupo holandés de Rotterdam, quienes establecieron los denominados criterios de Brighton(7)
Progresivamente se han descrito nuevos síndromes que se han sumado a las formas clásicas. Entre 2014 y 2019, surgieron diferentes clasificaciones que incluían todas las variantes clínicas, desde las formas clásicas hasta las más anecdóticas (Tabla 1), y en las que se propone como nomenclatura emplear el síndrome de Guillain-Barré como concepto “paraguas” indicando en un subíndice la variante clínica específica de la que se trata. Por ejemplo, “síndrome de Guillain-Barré, variante Miller-Fisher” (8) . No existen criterios clínicos cerrados para ninguna de estas variantes debido al grado de solapamiento entre ellas, lo que condiciona una predisposición hacia otras posibles formas de clasificación. Los estudios complementarios habituales sirven de apoyo al diagnóstico siempre que exista una sospecha fundamentada y sin ser diagnósticos en sí mismos.
Tabla 1 Variantes clínicas reconocidas del síndrome de Guillain-Barré y sus respectivas frecuencias estimadas (8).
| Variante | Frecuencia (% total del SGB) |
| Sensitivo-motora clásica | 30-85 % |
| Motora pura | 5-70% |
| Sensitiva pura | <1% |
| Paraparética | 5-10 |
| Faringo-braquio-braquial | <5% |
| Diparesia facial con parestesias | <5% |
| Síndrome de Miller-Fisher | 5-25 % |
| Sindrome Bickerstaff | <5% |
2. Hitos topográficos
Paralelamente a la descripción de las variantes sindrómicas han surgido conocimientos que cuestionan conceptos tradicionales. En primer lugar, la verdadera localización donde tienen lugar estos procesos.
En el caso de las variantes periféricas, existe un rechazo cada vez más patente a la categorización topográfica de los cuadros periféricos en desmielinizantes o axonales. En 1988 y 2015, Hadden y Rajabally respectivamente, proponían diferentes criterios para el diagnóstico neurofisiológico de las formas periféricas en desmielinizantes o axonales(9)(10) . Eran criterios estáticos, basados en un único estudio. Entre 2014 y 2018 Uncini y su grupo propusieron una clasificación basada en la verdadera localización donde ocurren estos fenómenos autoinmunes, los dominios nodales fundamentalmente(11‚ . El rechazo a la clasificación tradicional se basa en el hecho que los gangliósidos son parte de la mielina y del axón predominando en este último, dato que es conocido ya desde los años noventa. Conforman el anclaje entre ambos a nivel de los dominios nodales, donde se concentran fundamentalmente y donde ocurren realmente estos fenómenos. Siendo este escenario, la clasificación clásica de Hadden y Rajabally en formas axonales o desmielinizantes está abocada a caer en desuso, por ser incorrecta y porque ambos patrones neurográficos pueden estar justificados por una disfunción nodal-paranodal. Uncini y su grupo comenzaron a referirse a este grupo de entidades, en el que no únicamente se encuentran los síndromes antigangliósidos, en términos de “nodo-paranodopatías”. Asimismo, introdujeron el concepto de “fallo de conducción reversible”, estableciendo la necesidad de estudios seriados para establecer el pronóstico. Según este nuevo paradigma, los clásicos patrones axonales no tendrían por defecto un peor pronóstico, como se comprueba con frecuencia en la práctica clínica habitual. Por contra, la reversibilidad sería inversamente proporcional a la presión inmunológica y únicamente podría establecerse mediante sucesivos estudios(12) . Sin embargo, la realidad es que en la práctica clínica habitual estos conceptos no han sedimentado aún, y con frecuencia se sigue empleando la clasificación dicotómica de formas desmielinizantes o axonales.
N o hay que olvidar que existen signos clínicos en estos síndromes que no se explican únicamente por una disfunción nodal. Esto es debido a la presencia de gangliósidos a nivel del terminal presináptico de la placa motora y de los husos neuromusculares. En el primero de ellos, el ataque de los anticuerpos produce un fallo en la transmisión neuromuscular por la liberación masiva de cuantos de acetilcolina, es decir, por agotamiento. En 2009, Liu y su grupo proponían este mecanismo como principal responsable en la oftalmoparesia atribuida a estos síndromes, dada la alta concentración de gangliósidos en los terminales presinápticos de los nervios oculomotores con respecto a otros nervios (13)(14). En el caso de los husos, se presupone un bloqueo de la transmisión aferente como principal responsable de los cuadros de ataxia. Se trata de una hipótesis por exclusión, ya que no se ha podido demostrar origen cerebeloso, cordonal posterior o sensitivo para este síntoma hasta la fecha(15)
En el caso de las variantes centrales, se cuestiona el motivo por el que tienden invariablemente a la recuperación completa en comparación con las periféricas, en las que el 15% de los casos queda con secuelas, con el agravante de que la concentración de gangliósidos en el sistema nervioso central es tres veces mayor que en el periférico. Esto se atribuye a la alta eficiencia de la barrera hematoencefálica (BHE) con respecto a la hemato-nerviosa(16) . El área postrema se postula como área de filtración de los autoanticuerpos al sistema nervioso central por ser la BHE más vulnerable a este nivel. Este fenómeno viene estudiándose sobre todo en la neuromielitis óptica desde los años noventa, pero en la variante de Bickerstaff tan sólo se tiene evidencia parcial(17). Otra hipótesis acerca del acceso de los gangliósidos al sistema nervioso central es la de la endocitosis y transporte retrógado de los mismos a nivel del terminal presináptico de la placa motora. Este fenómeno fue propuesto por Cunningan y su grupo en 2016. (18)
3. Hitos inmunológicos
Pero no sólo cabe reconsiderar los conceptos topográficos hasta ahora expuestos. El segundo punto de inflexión surge de los conocimientos emergentes sobre inmunopatología. El entendimiento creciente sobre estos fenómenos autoinmunes nos predispone cada vez más hacia una clasificación centrada en los antígenos diana, los gangliósidos(19) . Los gangliósidos son una familia de glicoesfingolípidos ampliamente distribuidos por el sistema nervioso, donde su presencia es entre unas 10-30 veces mayor que en el resto de tejidos. Su nomenclatura está determinada por su composición estructural, determinada a su vez por los experimentos originales desarrollados por Svennerholm en 1980. Los gangliósidos están estructuralmente constituidos por una ceramida (ácido graso más esfingosina), un oligosacárido y un ácido siálico. Su nomenclatura está determinada por su composición estructural. La letra G se refiere simplemente a la sigla de su propio nombre, gangliósido, porque se aislaron por primera vez a nivel ganglionar. Se han descrito en torno a unos 200 subtipos diferentes aproximadamente hasta la fecha. Los sufijos M, D, T y Q indican el número de ácidos siálicos que contiene cada molécula (mono, di tri o tetra). Los sufijos numéricos son menos intuitivos. Hacen referencia a la longitud de la cadena glucídica que se une a la ceramida. Se rigen por la regla 5-X, donde X es el número de azúcares y será un número entero con signo positivo. Por ejemplo, si X es igual 4, los gangliósidos se denominarán GM1, GD1 o GT1; si X es igual a 3 los gangliósidos se denominarán GM2, GD2 o GT2; y si X es igual a 2 los gangliósidos se denominarán GM3, GD3 o GT3. Finalmente, el subíndice “b” se añade a aquellos gangliósidos con al menos un grupo disialosil (enlace de dos ácidos siálicos) unido a una galactosa interna. Por defecto, el subíndice “a” se añade cuando no hay tal grupo dialosil, sino que en du defecto, es un único ácido siálico el que se une a la galactosa (Figura 1) (20) .
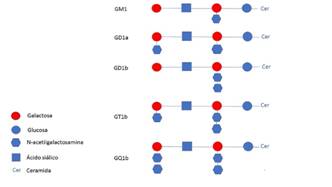
Los ácidos siálicos son el elemento diferencial de los gangliósidos con respecto a otros glicoesfingolípidos. Deben su nombre al término “saliva” porque se identificaron por primera vez en la mucina salivar bovina en 1955. Son azúcares de nueve carbonos cargados negativamente que se unen lípidos y proteínas. Se han descrito más de 80 tipos de ácidos siálicos en la naturaleza. Sin embargo, elácidoN-acetilneuramínico (Neu5Ac) y elácidoN-glicolilneuroamínico (Neu5Gc) son, con diferencia, los ácidos siálicos más abundantes en el sistema nervioso. Su función dentro de la estructura de los gangliósidos es divergente. Por un lado, proveen de una sólida protección contra agentes microbiológicos externos gracias a que sus cargas negativas se exponen en la superficie de la membrana repeliendo la unión de células o macromoléculas. Por contra, algunos virus, bacterias y parásitos han aprendido a explotarlos como sus receptores. (20) .
Los gangliósidos-like son moléculas que guardan similitud estructural con los gangliósidos. Se encuentran fundamentalmente en la superficie de microorganismos. De todos los agentes patógenos implicados en estos síndromes, el mejor conocido es el C. jejuni, por ser el más frecuentemente responsable (en torno al 40% de los casos), y aquél en cuya investigación más se ha profundizado. Hay que destacar, a tal efecto, los innumerables trabajos del profesor Yuki y su grupo, llevados a cabo desde finales de los años ochenta hasta la fecha. Fueron pioneros en proponer una nomenclatura estructurada en dos niveles: 1) el clínico, basado en las diferentes variantes sindrómicas, y 2) el inmunológico, basado en los autoanticuerpos contra los antígenos diana. En cuanto a las vacunas como moléculas gangliósidos-like, tan solo la de rabia inactivada y la de la gripe A han demostrado formalmente guardar una relación causal con estos síndromes. Queda esperar a los resultados de estudios en curso para el caso de la vacuna contra el SARS-CoV2. (21)(22)
Los anticuerpos antigangliósidos fueron descritos por primera vez en el encéfalo de ratones en 1972. Su presencia se mide en suero, aunque el LCR puede contener pequeñas cantidades procedentes de la circulación sistémica. (12)
La teoría fundamental sobre la fisiopatología de los síndromes antigangliósidos está consensuada por la mayoría de los autores ya desde los años noventa. Se produce una reacción cruzada entre los gangliósidos y las moléculas gangliósidos-like, de manera que los autoanticuerpos se unen a los gangliósidos propios desencadenando la activación de la vía clásica del complemento y la destrucción local(23)(24) .
A lo largo de sucesivas décadas, se ha desentrañado paulatinamente el papel de cada gangliósido por separado. Hoy conocemos que cada gangliósido tiene una distribución más o menos definida en el sistema nervioso (Tabla 2) (25) . Ello acota el espectro clínico para cada autoanticuerpo en concreto, no exento de cierto grado de solapamiento (Tabla 3) (26)(27) .
Tabla 2 Localización principal de los gangliósidos asociados a los síndromes antigangliósidos (25).
| Gangliósido | Principal localización |
|---|---|
| GM1 | Sustancia blanca, hipotálamo, raíces dorsales, terminal presináptico placa motora |
| GD1a | Sustancia gris, bulbo olfatorio, sustancia negra, terminal presináptico placa motora |
| GD1b | Sustancia gris, sustancia blanca medular, epitálamo |
| GT1a | Dominios nodales (axón y mielina) |
| GT1b | Globus pallidum, raphe magnus |
| GM3 | Endotelio |
| GQ1b | Dominios nodales (axón y mielina), terminal presináptico placa motora |
Tabla 3 Síndromes neurológicos asociados a los diferentes gangliósidos en su forma simple o en forma de complejos (25)(26).
| Síndrome antigangliósido | Gangliósido o complejo gangliósido asociado |
|---|---|
| Neuropatía idiopática desmielinizante aguda (de sus siglas en inglés, AIDP) | GT1b (50% de los casos) |
| Neuropatía motora axonal aguda (de sus siglas en inglés, AMAN) | GM1, GM1a, GD1a, Anti-GM1GalNAc-GD1a, Anti-GM1/GA1 |
| Paraparética | GM1, GM1a, GM1b, GD1a, GD1b |
| Neuropatía sensitivo-motora axonal aguda (de sus siglas en inglés, AMSAN) | GM1, GM1a, GM1b, GD1a |
| Faringocervicobraquial | GT1a |
| Diparesia facial con/sin parestesias | GT1a |
| Sensitiva pura | GD1b, GM2 |
| Miller-Fisher | GQ1b, GT1a |
| Síndrome de Bickerstaff | GQ1b |
No obstante, aunque disponemos de una teoría globalmente aceptada para estos síndromes, existen otros muchos factores también integrantes en su fisiopatología cuyo peso específico resta aún por delimitar. Únicamente destacar el novedoso papel de los complejos de gangliósidos, el cual cobra cada vez más relevancia en las diferentes líneas de investigación. La conformación de estos complejos genera nuevos epítopos que inducen una mayor, o excepcionalmente menor, reactividad inmunológica que cada gangliósido en sí mismo de forma aislada. La dificultad para detectar estos complejos radica es que se requiere de glycoarrays, los cuales son capaces de identificar complejos de anticuerpos en un 53% de los casos con respecto al 13% detectable mediante técnicas habituales. Los complejos que mayor reactividad provocan están conformados por GQ1b o GT1a junto con algún otro gangliósido. Se cree que este es el principal mecanismo implicado en las denominadas “formas desmielinizantes” en las que no se ha demostrado la implicación de un gangliósido específico aislado(26).
4. Hitos terapéuticos
Las terapias empleadas para el tratamiento de estos síndromes también han sufrido una evolución paralela al conocimiento de su fisiopatología. El recambio plasmático se empleó por primera vez en 1959 en el tratamiento de la púrpura trombocitopénica y en 1978 en el del SGB. Posteriormente, se confirmaría su eficacia mediante ensayos clínicos controlados (nivel de evidencia A, clase II). Su efecto terapéutico se atribuye a la eliminación de inmunoglobulinas, interleuquinas, citoquinas y otros factores proinflamatorios circulantes como los del sistema del complemento. (28).
Las inmunoglobulinas intravenosas se emplearon por primera vez en el SGB en 1988. Su equivalencia en cuanto a eficacia con respecto al recambio plasmático se evidenció en 1992 (nivel de evidencia A, clase I). Se hipotetiza que las inmunoglobulinas modulan el sistema inmune de diversas maneras: inhibiendo la vía del complemento, modulando los linfocitos T, suprimiendo el papel de los macrófagos, etc. Si bien, la razón última por la que estos dos tratamientos son eficaces no está bien establecida. En la actualidad, las inmunoglobulinas intravenosas son la primera línea de tratamiento en los países desarrollados por su menor tasa de efectos secundarios con respecto al recambio plasmático. Ensayos clínicos en los que se combina tratamiento con inmunoglobulinas y recambio plasmático han fracasado a la hora de demostrar su beneficio en relación con cada uno de estos tratamientos de forma independiente(28).
Los corticoides se usaron por primera vez en 1952 con aparente éxito entonces, manteniéndose su uso durante casi dos décadas. Posteriormente se llegó a la conclusión de que eran ineficaces para el tratamiento del SGB, desaconsejándose formalmente su uso (nivel de evidencia A, clase I) (28).
Las terapias centradas en el sistema de complemento como principal elemento inmunológico implicado conocido han sido también objeto de desarrollo. El mensilato de nafamostat se ha propuesto como tratamiento del SGB. Se ha usado en Japón como terapia contra la coagulación intravascular diseminada o la pancreatitis aguda sin efectos adversos reseñables. Se trata de un inhibidor sintético de las serinproteasas del sistema del complemento, principal responsable del daño nervioso en estos procesos. Sin embargo, esta propuesta no ha contado con el apoyo de la industria farmacéutica hasta la fecha y no ha continuado desarrollándose(29). Los anticuerpos monoclonales contra el sistema de complemento anti-CR1 (mirococept) y anti-C5 (eculizumab) se probaron sin éxito en un modelo murino. Actualmente, existe un ensayo clínico en fase II en torno al anticuerpo monoclonal ANX005 (inhibidor de C1q) administrado en combinación con inmunoglobulinas endovenosas. La situación actual de este ensayo es que faltan por aclarar aspectos con relación a la combinación terapéutica y modificaciones de dosis para poder proseguir con el mismo(30).
Ha habido tímidos intentos de emplear otros fármacos inmunomuduladores como son el interferón alfa, la azatriopina, el metrotexate, la ciclosporina, el rituximab o el alentuzumab. Ninguno de ellos ha demostrado eficacia significativa, por lo que no se consideran una alternativa terapéutica(28) . Recientemente se están proponiendo nuevas dianas terapéuticas que tienen por objetivo limitar el tránsito de leucocitos a través de la BHE y hemato-nerviosa. Existen varios estudios incipientes en marcha, los cuales aún se están desarrollando en modelos animales(31)(32)
Conclusiones
Como en otros ámbitos de la neurología, es necesario llevar a cabo revisiones periódicas del estado de conocimiento de un tema conforme se amplían las investigaciones al respecto. Se trata de ser rigurosos en los conceptos que se manejan habitualmente.
En el presente trabajo, se revisa la evolución histórica de los síndromes antigangliósidos y se actualizan conceptos y se plantea la necesidad de llevar a cabo una nueva clasificación estratificada en dos niveles: clínico e inmunológico. Puede que en un futuro a medio plazo comencemos a hablar en términos de “síndrome de Guillain-Barré, variante Miller-Fisher” o “síndrome de Guillain-Barré, variantes anti-GQ1b y GT1a”
Serán necesarias ulteriores consensos para unificar conceptos y avanzar hacia terapias más efectivas en el futuro.