









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Wilson (EW) es una tesaurismosis hereditaria autosómica recesiva que deteriora el metabolismo del cobre; en el ser humano la ingesta de este metal (1 mg diario) excede los requerimientos normales por lo que la única manera de homeostasis es su excreción mediante la bilis (90%) y por la orina (10 %) (1). Se han detectado hasta 600 anomalías diferentes en el gen ATP7B, localizado en el cromosoma 13, que codifica una proteína transportadora en la membrana celular del hepatocito encargada de la liberación del cobre en la bilis, y que además proporciona este ion a la ceruloplasmina (la principal proteína transportadora del cobre en el organismo) (2). El mecanismo fisiopatológico subyacente a la enfermedad de Wilson -así denominada en honor a Samuel K. Wilson, quien describió la primera serie de casos en la literatura moderna en el año 1912- es la acumulación de cobre en el hígado y posteriormente en órganos como el cerebro, riñones, córnea, corazón, entre otros, donde produce daño mediante estrés oxidativo y generación de radicales libres (1)(3). Aún se desconoce por qué algunas estructuras encefálicas como ganglios basales, tálamo, cerebelo y tallo cerebral son más susceptibles a la toxicidad por cobre (astrogliosis, desmielinización y desintegración tisular) y donde la concentración cúprica llega a ser 10 a 15 veces mayor que en sujetos control (4).
La prevalencia de la enfermedad de Wilson oscila entre 1 en 30 000 a 1 en 100 000 y la frecuencia de portador de la anomalía genética subyacente actualmente se cifra en 1:50 (1). Hay serias evidencias de que la EW podría ser más frecuente, pero subdiagnosticada (5). La edad de inicio suele ser entre la infancia y la adultez joven (5 a 35 años) aunque se han reportado casos desde los tres a más de setenta años de edad. En cuanto a la expresión clínica, existe creciente evidencia de que la EW puede presentarse durante años exclusivamente con síntomas psiquiátricos (2) mientras que la implicación hepática y neurológica puede resultar evidente solo después de algunos años, con el consiguiente retraso del diagnóstico. Las manifestaciones tempranas de la EW son hepáticas en alrededor de 50% de los casos, neurológicas en 40% y psiquiátricas en 20% de los casos pero hay una variabilidad amplia, más infrecuentes son las presentaciones tempranas con síntomas predominantemente hematológicos, renales, osteoarticulares, etc. (1). Un 30% de casos son asintomáticos y se detectan en el tamizaje de familiares de personas afectadas. En la medida que progresa la evolución clínica, hasta 100 % de casos pueden llegar a presentar síntomas psiquiátricos (6). En general, los síntomas neurológicos y psiquiátricos suelen estar inextricablemente unidos en su presentación (7).
El diagnóstico correcto de la enfermedad de Wilson solo sucede en un tercio de los casos al momento de su presentación (8) y hasta 20% de los pacientes con EW han visitado a un psiquiatra antes de su diagnóstico firme (1). Los síntomas psiquiátricos poseen valor pronóstico, revelan una patología más severa o avanzada, y aunque hay diferentes cursos evolutivos -aún con tratamiento- un diagnóstico precoz puede evitar secuelas permanentes y hasta la muerte, como en pocas patologías genéticas (2).
A propósito de un caso de EW en que el diagnóstico no fue precoz, y la paciente sufrió secuelas irreversibles, revisamos la literatura actual para enfatizar, pese a la rareza de estos casos clínicos, la necesidad de un afronte sistemático e integral de los cuadros de presentación psiquiátrica desde un punto de vista holísticamente neuropsiquiátrico.
Caso clínico
Paciente mujer de 36 años de edad, soltera, instrucción secundaria, natural y procedente de Lima, sus padres eran primos hermanos. Inició su enfermedad a los 18 años de edad con aislamiento, mutismo, lloraba a escondidas, pero al ser preguntada sobre esto, ella refería que no le pasaba nada. Luego de casi un año su ánimo triste fue más evidente: expresaba ideas de minusvalía y se reprochaba ser “una carga para su familia” por haber abandonado sus estudios superiores. En los 4 meses siguientes su apetito disminuyó y bajó de peso. Luego de una riña doméstica intrascendente, la encontraron en el baño con una botella de ácido muriático pero le impidieron beberla. Recibió atención psiquiátrica de emergencia y manifestó que meses atrás había intentado suicidarse tomando 20 tabletas de paracetamol e ingiriendo raticida. También refirió alucinaciones auditivas imperativas: “suicídate”, “mátate”, y delirios de referencia: creía ser constantemente observada en la calle. Se le diagnosticó depresión con síntomas psicóticos y se le prescribió fluoxetina 20 mg, risperidona 2 mg y clonazepam 1 mg diarios.
A la semana de tratamiento presentó rigidez muscular, temblor distal en dedos de las manos e hipocinesia. Atendida en emergencia, le indicaron biperideno 5 mg IM, y se cambió la risperidona por sulpirida 200 mg diarios, sin embargo, los síntomas extrapiramidales persistieron sin variación. En controles sucesivos la paciente continuó presentando alucinaciones auditivas imperativas de suicidio, marcha rígida, mirada fija, hipomimia y bradicinesia. El planteamiento diagnóstico fue un trastorno depresivo con síntomas psicóticos versus trastorno esquizofreniforme, se le indicó risperidona 2 mg, mirtazapina 30 mg y clonazepam 2 mg diarios.
Sin embargo, los signos extrapiramidales y los síntomas psicóticos y depresivos -incluyendo ideación suicida- se mantuvieron sin mayor cambio. Lucía sialorreica, disártrica, con prominente parkinsonismo que le dificultaba hacer sus actividades cotidianas como vestirse y asearse. Los síntomas extrapiramidales tenían leves fluctuaciones, disminuían parcialmente al retirarse la risperidona pero al recrudecer los síntomas psicóticos y reiniciarse este antipsicótico, reaparecían con intensidad.
Dos años después del inicio de sus síntomas (20 años de edad), no se observaban cambios clínicos positivos: ahora presentaba también alucinaciones visuales y delirios de embarazo, exhibía conducta pueril y sus síntomas motores eran más ominosos: tenía disfagia, distonía de músculos masticatorios y bradicinesia, pese al aumento de la dosis de biperideno a 6 mg por día y la rotación a otros antipsicóticos como clorpromazina 50 mg u olanzapina 5 mg diarios.
La paciente fue hospitalizada finalmente por presentar conducta desorganizada, dificultad para deglutir y deambular y disminución de peso. Se planteó el diagnóstico de esquizofrenia paranoide, inicialmente solo se le indicó biperideno y diazepam condicional, se suspendió la olanzapina y se consideró la posibilidad de terapia electroconvulsiva (ECT). Sin embargo, a pesar de recibir anticolinérgicos y no antipsicóticos, se observó intensificación de la disartria: solo podía comunicarse escribiendo; también aumentaron la sialorrea y el tremor digital. En ese momento se planteó el diagnóstico de síndrome parkinsoniano de etiología a determinar. Se solicitó evaluación neurológica, pruebas de función hepática y estudio de imágenes cerebrales. Solo se indicó trihexifenidilo 15 mg/día.
La paciente empezó a recibir ECT bitemporal, tres sesiones por semana: inicialmente hubo aparente mejoría de la sialorrea y también de la rigidez, luego de la quinta sesión la disartria mejoró y desaparecieron los delirios y alucinaciones Se llegó a 10 sesiones de ECT, con mejoría de síntomas psicóticos y parkinsonianos. Se agregó como antipsicótico quetiapina 25 mg/día, y progresivamente se fue incrementando la dosis.
Al cabo de una semana se retomaron sesiones de ECT dos veces por semana debido a la reaparición de lenguaje desorganizado, disartria y rigidez; se suspendió la quetiapina (100 mg/día), y se indicó nuevamente trihexifenidilo 5 mg/día; después de lo cual se evidenció mejoría parcial de la disartria, pero la rigidez persistió igual y la paciente negó la presencia de síntomas psicóticos. Se completaron 4 sesiones de ECT adicionales: en total la paciente recibió 14 sesiones.
Ante las alteraciones motoras como parkinsonismo, rigidez muscular generalizada, disfagia y disartria, el equipo médico de hospitalización planteó la posibilidad de un problema orgánico no sospechado, por ejemplo, enfermedad de Wilson o síndrome de Hallervorden-Spatz. En el examen físico se verificó hipotrofia muscular en manos y dedos en garra en los dos primeros dedos de la mano izquierda, pero no se evidenció hepato ni esplenomegalia. Dos semanas después, la interconsulta a oftalmología informó presencia de anillos de Kayser-Fleischer, lo que orientó definitivamente el diagnóstico hacia enfermedad de Wilson. Se coordinó transferencia a servicio de neurología de hospital general y se realizó modificación de la dieta a baja en cobre. Al cumplir el 62° día de hospitalización llegó resultado de ceruloplasmina baja (4,8 mg/dL), y se inició tratamiento quelante con D-penicilamina 250 mg cada 8 horas. Es decir, el diagnóstico firme de enfermedad de Wilson fue establecido casi tres años después del inicio de los síntomas.
Luego de seis años de enfermedad, a la edad de 24, y con tratamiento irregular, en la paciente se evidenciaba afasia de expresión (no emitía palabras, solo algunos sonidos), se comunicaba por gestos, mostraba déficit del equilibrio y la marcha por debilidad marcada en las cuatro extremidades. Se sugirió inicio de rehabilitación física.
Actualmente, con 37 años de edad, ya no recibe penicilamina por no hallarse disponible en el país, sino sulfato de zinc. Ha dejado de recibir rehabilitación física por la pandemia de Covid y eso ha determinado que presente graves limitaciones para sus actividades de la vida diaria. Presenta muy severa disartria y puede caminar pocos pasos. Requiere atención permanente de la familia.
DISCUSIÓN
La enfermedad de Wilson es una de las escasas enfermedades congénitas que pueden ser completamente tratables si se detectan a tiempo (3); a diferencia de la mayoría de enfermedades hereditarias raras, los tratamientos disponibles permiten sobrevivencias mayores a 90%, y el pronóstico es excelente en aquellos casos tratados tempranamente (1). Nuestro caso es manifestación de un retraso diagnóstico que acarreó daños neurológicos irreversibles al punto de constreñir a nuestra paciente a una vida muy limitada en su funcionalidad y autonomía. El retraso diagnóstico reportado en la bibliografía excede los dos años y es aún mayor cuando la presentación es preferentemente psiquiátrica (4). Esto puede atribuirse a causas muy diversas (9): la gran variabilidad de los signos y síntomas -se ha llamado a la EW “el gran enmascarado”- (10), la baja incidencia de la patología, la valoración sesgada de los datos clínicos, el escaso índice de sospecha y el desconocimiento de esta enfermedad por los evaluadores, entre otros (1)(2)(4). Como establecía T.R. Dening, estudioso de la EW, no se espera que el psiquiatra sea capaz de diagnosticar esta patología, pero es imprescindible su sospecha diagnóstica adecuada (7). El proceso diagnóstico de la EW, de hecho, es complejo y exige amplia experiencia clínica y adecuada interpretación de las ayudas diagnósticas (ver Tabla 1)-pues no hay una prueba única que diagnostique la EW- (11)(12).
Tabla 1 Hallazgos de laboratorio y clínicos usados en el diagnóstico de EW (12).
| 1. Niveles de ceruloplasmina sérica por debajo del 50% del valor normal (< 0,2 g/L). 2. Incremento de la excreción urinaria de cobre en 24 horas (> 1,6 mmol / 24 h en adultos y > 0,64 µmol / 24 h en niños. 3. Cobre libre en plasma > 1,6 mmol / L. 4. Cobre hepático > 4 µmol / g de tejido deshidratado. 5. Presencia de anillo de Kayser-Fleischer en la córnea, examinado con lámpara de hendidura por oftalmólogo con experiencia. |
Aunque las manifestaciones clínicas predominantes de la EW son las hepatopatías y los cuadros neuropsiquiátricos, no hay signos clínicos patognomónicos para el diagnóstico y no hay dos pacientes con cuadros clínicos iguales, aún dentro de una misma familia (3). Tampoco se ha logrado adscribir cuadros clínicos específicos a determinadas anomalías genéticas. La penetrancia incompleta, las múltiples mutaciones involucradas y factores epigenéticos diversos, explican de alguna manera este peculiar panorama clínico (3). Es decir, la EW es una enfermedad clínica y genéticamente muy heterogénea (11).
Las formas de presentación hepática de la EW suelen comenzar antes, al acabar la infancia o en la adolescencia, y abarcan desde formas asintomáticas de elevación de enzimas transaminasas hasta fallas hepáticas fulminantes y cirrosis crónica. Se ha sugerido que cualquier hepatopatía de origen desconocido en individuos jóvenes se considere como probable EW hasta que se demuestre lo contrario (9). Las formas neurológicas suelen acontecer a partir de la edad de 20 años y sus síntomas principales son temblor intencional y postural -usualmente burdo, asimétrico y de tipo “aleteo”- (80%), rigidez y parkinsonismo (40%), distonías focales o generalizadas (10-30%) (1)(2). Entre 70 a 100% de pacientes con EW manifiestan síntomas psiquiátricos, los que pueden estar presentes desde el inicio y anteceder a todo el cuadro clínico (2), o manifestarse durante el curso de cuadros neurológicos, hepáticos o mixtos, o representar la respuesta psicológica a una patología crónica y gravosa, o incluso pueden ser efectos secundarios del tratamiento (1). El porcentaje de casos de EW que empiezan solo con síntomas psiquiátricos es muy variable según las series (desde 1% a 30%) e igualmente a las formas de inicio neurológico, principian en la segunda década de la vida (1)(4)(6). Frecuentemente los síntomas psiquiátricos iniciales son inespecíficos y poco discernibles con avatares propios de la adolescencia: merma del rendimiento escolar, impulsividad, conducta inapropiada (6).
Las manifestaciones psiquiátricas pueden clasificarse de diferentes modos (ver Tabla 2):
Tabla 2 Síntomas neuropsiquiátricos frecuentes en EW (7).
| 1. Disturbios de la personalidad y la conducta: - Agresividad, rebeldía, impulsividad, descuido de las labores escolares, irresponsabilidad, consumo de sustancias psicoactivas. 2. Disturbios afectivos: - Labilidad emocional, irritabilidad, ideas e intentos suicidas, síndrome depresivo, síndrome maniaco. 3. Disturbios cognitivos: - Bradipsiquia, déficit de memoria, errores de juicio. 4. Disturbios psicóticos: - Delirios, catatonía, alucinaciones (raras). |
Hay que reconocer que a veces un mismo hallazgo (p.ej.: irritabilidad) puede ser adjudicable al ámbito de lo afectivo o de la personalidad por lo que la consideración exploratoria ha de ser amplia y desprejuiciada. Debe evitarse la precipitada “explicación psicológica” acerca de que los síntomas afectivos son siempre secundarios al sufrimiento del paciente por su dolencia: se ha demostrado la presencia de síntomas depresivos desde estadios muy tempranos, sin ningún grado de deterioro en el paciente, lo que contradice tal hipótesis. Y además se ha demostrado, con neuroimágenes, el origen orgánico cerebral de estos cuadros depresivos (1).
Debe recordarse que, en los estudios últimos, controlados y con instrumentos estandarizados, las manifestaciones afectivas (síndromes depresivos, maniacos y mixtos, y crisis suicidas) son más frecuentes que los síntomas psicóticos (1). Ha sido un sesgo de series más tempranas, al no usar instrumentos estructurados, el diagnóstico más frecuente de cuadros de tipo esquizofreniforme. (1). Incluso algunos estudios epidemiológicos recientes encuentran similar incidencia de psicosis en la población general y en los que padecen EW (4) El síndrome depresivo puede hallarse en 60% de los pacientes con EW y también una alta tasa de crisis suicidas (hasta 16% de los pacientes) (4). Actualmente los síntomas psicóticos se consideran poco frecuentes y hasta raros (11), pero cuando se presentan pueden semejar una típica esquizofrenia paranoide con prominentes signos extrapiramidales asociados (11)(13), y es frecuente que se los confunda como secundarios a los antipsicóticos prescritos. Ahora bien, es cierto que no pueden distinguirse clínicamente los síntomas psiquiátricos de la EW de aquellos propios de un trastorno psiquiátrico primario, por ello algunos autores, asumiendo que la frecuencia de causas orgánicas en un primer episodio psicótico oscila alrededor de 3%, sugieren un tamizaje obligatorio de EW en todos los primeros episodios de psicosis (4).
El anillo de Kayser-Fleischer consiste en una coloración dorada o verdosa en la periferie de la córnea, debida a la acumulación de cobre en la membrana de Descemet, y que se presenta hasta en 90 a 99% de casos de EW con manifestaciones neurológicas, mientras que en las formas hepáticas la frecuencia es apenas alrededor de 50% (14). Es preferible evaluarlo con lámpara de hendidura para su adecuado diagnóstico. Hay que señalar que no es patognomónico de EW pues también puede apreciarse en cirrosis biliar primaria o hepatitis crónica activa, entre otros diagnósticos (14). (Figura 1)
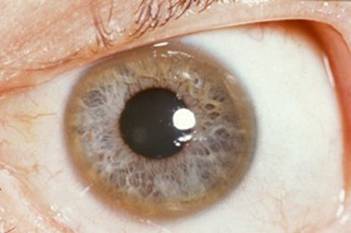
Aunque las neuroimágenes no están consideradas dentro de los criterios establecidos para el diagnóstico de la EW, debe recordarse que la resonancia magnética de encéfalo es siempre anormal en EW de presentación neurológica (señales hiperintensas en T2 de ganglios basales, mesencéfalo y cerebelo) pero puede ser normal en los casos hepáticos. Las lesiones de sustancia blanca y corteza son infrecuentes (9).
Nuestro caso es paradigmático por la edad de inicio juvenil y con presencia de síntomas depresivos, psicóticos y crisis suicidas. No se efectuó una exploración neurológica amplia desde las primeras consultas -es probable que los síntomas neurológicos estuviesen ya presentes, pero recién se advirtieran al recibir psicofármacos, con tremor, rigidez e hipocinesia-, sin embargo, tampoco se hizo tal evaluación durante la evolución, en que equivocadamente se asumía que el parkinsonismo era solo secundario a los antipsicóticos, pese a las reducidas dosis o a su persistencia aparte de la medicación. Se ha señalado que debe considerarse el diagnóstico de EW en toda persona joven con alteraciones de la fonación y en el uso de las manos (8). Asimismo, está registrada la confusión que puede generar la presencia de parkinsonismo prominente, erróneamente atribuido a los antipsicóticos. El mecanismo patogénico subyacente es una disrupción en la transmisión dopaminérgica (la densidad de receptores D2 se reduce en el estriado). Lastimosamente, el uso de antipsicóticos como haloperidol y otros antipsicóticos típicos, pero también risperidona (6) se asocia a un riesgo de mayor deterioro neurológico, por lo que deben ser usados, de ser necesario, con el máximo cuidado, a la menor dosis y el tiempo mínimo indispensable, dando prioridad a aquellos con menor efecto extrapiramidal y menor metabolismo hepático (sulpirida y amisulprida son destacables). Respecto a la terapia electroconvulsiva (ECT) en EW, hay reportes de caso que demuestran su eficacia en cuadros depresivos, catatónicos, maniacos y también psicóticos, sobre todo cuando se deben evitar los antipsicóticos o en casos de severa desorganización o estupor catatónico, aunque en nuestro caso, y dado que aún no se iniciaba la terapia específica contra EW, su utilidad fue limitada. El estándar de oro del tratamiento de la EW sigue siendo la terapia quelante que disminuye los niveles de cobre en el organismo (penicilamina, trientina, molibdato de amonio) o inhibe su absorción (sales de zinc) (13).
Aparte de las limitaciones inherentes a cualquier reporte de caso, debe señalarse aquí la carencia de neuroimágenes ilustrativas y el seguimiento irregular que impidió mayor abundancia de datos clínicos, incluyendo el estado actual de nuestra paciente con mayor detalle.
En conclusión, dada la polimórfica presentación que difumina un cuadro clínico característico, y las ominosas consecuencias de no ser detectada a tiempo, la EW debiera ser considerada en el diagnóstico diferencial de todo paciente joven con un primer episodio de psicosis. La hipersensibilidad marcada a los efectos secundarios de tipo parkinsonismo, con prominente disartria, rigidez y sialorrea ante el uso de antipsicóticos debe ser un llamado de alerta inobjetable para considerar una posible EW (16). Es importante que los profesionales “piensen” en esta enfermedad para poder detectarla.