









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Joubert (SJ) (OMIM 213300) es una malformación congénita autosómica recesiva clasificada dentro de las Ciliopatías genéticas. Existen alrededor de 13 genes asociados: AHI1, CPLANE1, CSPP1, INPP5E, KIAA0586, MKS1, TCTN2, NPHP1, CEP290, TMEM67, TMEM 216 RPGRIP1L, ARL13B y CC2D2A. Estos codifican proteínas estructurales relacionadas con los cilios primarios que cumplen funciones en los fotorreceptores, proliferación neuronal, células del túbulo renal y conductos biliares(1)(2). A su vez, cumplen funciones en el desarrollo del cerebelo, vermis y tallo encefálico, regulando algunas vías embrionarias implicadas en procesos de proliferación del neuroblasto, en la migración, diferenciación de las células de Purkinje y neuronas granulares(2)(3)(4).
Esta condición fue descrita en 1969 en 4 individuos de una misma familia que presentaban agenesia del vermis cerebelar, apnea-hiperpnea, ataxia, movimientos oculares anormales y retraso psicomotor(5). La prevalencia de este síndrome se encuentra entre 1:80.000 a 1:100.000 nacidos vivos(2)(6)(7) que puede estar subestimada(6). Clínicamente, se identifica desde la etapa neonatal con trastornos en el ritmo respiratorio, nistagmus y alteraciones en la deglución; posteriormente predomina la hipotonía, la ataxia y la discapacidad intelectual(7). Al examen físico se encuentra: frente prominente, epicanto, ptosis palpebral y baja implantación de pabellones auriculares(1). Otras manifestaciones que se pueden encontrar son: tubulopatía renal, inmunodeficiencia y, a nivel cerebral, trastornos de la migración neuronal en tronco encefálico(7).
En la resonancia magnética nuclear cerebral se han descrito, en el corte axial, la elongación de los pedúnculos cerebelosos superiores, displasia cerebelosa y fosa interpeduncular ensanchada o “signo del molar”, el cual está dado por la ausencia de las fibras decusadas del tracto peduncular superior del cerebelo(8)(9). Pueden presentarse alteraciones de forma extensa y difusa en los hemisferios cerebelosos, el vermis poco desarrollado dentro de una fosa posterior pequeña, el cuarto ventrículo en comunicación con una gran cisterna magna, hipoplasia del tronco encefálico y agenesia del cuerpo calloso(8). Algunos autores han propuesto clasificaciones clínicas donde se correlacionan el genotipo-fenotipo con las manifestaciones clínicas y la imagenología(7).
Presentamos este reporte con el fin de dar a conocer el caso de un paciente adulto con SJ, las características con las cuales se estableció su diagnóstico, complicaciones y resaltar un problema para tener en cuenta en estos pacientes: los trastornos del sueño en la vida adulta. Al momento de la revisión, este era el primer caso reportado con SJ en Colombia.
Caso clínico
Paciente de 39 años de sexo masculino, diestro, natural y procedente de Pereira en Risaralda - Colombia, con discapacidad intelectual de severidad indeterminada. Acude al Servicio de Neurología, acompañado de su madre, por presentar un cuadro clínico de varios años de evolución, caracterizado por movimientos incesantes de los miembros inferiores al dormir acompañado de ronquidos ocasionales no perturbadores, pausas respiratorias y somnolencia diurna excesiva. Realiza siesta de aproximadamente 60 minutos.
Antecedentes personales: Segundo hijo, nacido a término por vía vaginal y sin complicaciones; la madre refiere llanto demorado y retraso en el desarrollo psicomotor. Paciente que no tuvo acceso a servicios educativos, pero sabe leer y escribir su nombre. Antecedentes patológicos: hernia hiatal, asma y enfermedad por reflujo gastroesofágico. Farmacológicos: Montelukast 10 mg cada noche, Loratadina 20 mg cada 12 horas y Omeprazol 100mg al día.
Examen físico: presión arterial de 110/70 mmHg, frecuencia cardíaca a 72 latidos por minuto, peso de 63.3 Kg, talla de 1.66 m, IMC de 23.05 Kg/m2 y circunferencia del cuello de 40 cm. Obtuvo una puntuación de 9/24 en la escala de Epworth. Paciente con habla disártrica, falla de memoria a corto plazo y alteración en la semántica. Función del lenguaje está conservada.
Paraclínicos: Al paciente se le realizó una polisomnografía basal con oximetría de pulso (Tabla 1) la cual fue interpretada según los criterios de la Academia Americana de Sueño(¡0). Se estableció el diagnóstico de Síndrome de Apnea Central del Sueño del Adulto. Se sugirió ampliar estudios para precisar la etiología de este trastorno respiratorio y la realización de un estudio de titulación de presión aire positivo.
Tabla 1 Parámetros de la polisomnografía del paciente adulto con apnea central de sueño y síndrome de Joubert.
| Arquitectura del sueño | |
| Variable | Valor |
| Tiempo total registro (min)1 | 431 |
| Tiempo total sueño (min) | 333 |
| Eficiencia sueño (%) | 77,26 |
| Inicio de MOR2 (min) | 168 |
| Etapa N1 (%) | 6 |
| Etapa N2 (%) | 81 |
| Etapa N3 (%) | 7 |
| MOR (%) | 6 |
| Movimiento total de miembros | 15 |
| Eventos respiratorios | |
| Variable | Valor |
| Apnea central | 128 |
| Hipopnea central | 0 |
| AHI3 central | 22,8 |
| Apnea obstructiva | 1 |
| Promedio saturación de oxígeno | 92% |
*1 min: minutos. 2MOR: movimientos oculares rápidos. 3AHI: Índice apnea-hipopnea.
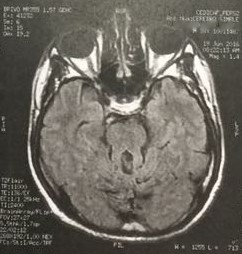
En imagen por Resonancia Magnética se describe un “aspecto hipoplásico de los lobulillos cefálicos del vermis y de los hemisferios cerebrales, adoptando el signo del molar, cambios displásicos en el cuerpo calloso con aspecto engrosado de su cuerpo e hipoplasia del pico”; por ende, se debe descartar síndrome de Joubert (Figura 1).
Discusión
En el caso clínico se describe un paciente diagnosticado de SJ en la edad adulta por medio de imagenología y estudios paraclínicos del sueño. El diagnóstico se basa en la clasificación propuesta por Brancati et al. y se describió un SJ puro, catalogado como una patología de mutación poligénica(7). Para establecer cuál de los 6 subtipos presenta el paciente, es necesario estudiar mutaciones genéticas puntuales, cuya descripción excede los límites de esta presentación.
Resaltar que el paciente en cuestión ha convivido con alteraciones del sueño y de la respiración, contrario a lo descrito en la literatura que indica que las alteraciones respiratorias mejoran con la edad del paciente(7)(11). Otros reportes en población pediátrica han resaltado que estas alteraciones del sueño se han asociado a diferentes síndromes, especialmente en el SJ, se incluye la apnea central del sueño de forma periódica que empeora en fases no MOR. Por ende, se sugiere control con polisomnografía desde los 12 meses de edad(12).
Un paciente de 15 años en Japón fue diagnosticado con SJ y presentó somnolencia diurna y alteración del patrón respiratorio relacionado con el sueño. Le realizaron una polisomnografía donde identificaron apnea de tipo central con un índice apnea-hipopnea de 16 eventos/hora. Un año después, con terapia suplementaria de oxígeno, mejoró su condición de base(11). Otro estudio, reportó un individuo de 25 años con diagnóstico tardío de SJ, apnea central severa y respiración periódica con saturación baja. Fue tratado con BiPAP en modo ST, con mejoría en su calidad de sueño y saturación de oxígeno(9). Estos casos coinciden en algunos de los hallazgos de nuestro paciente como lo son: la edad tardía en el diagnóstico del síndrome de Joubert, la apnea central del sueño explicada por una actividad refleja del tronco cerebral y que llevó al uso de dispositivos externos para mejorar la capacidad respiratoria y el sueño.
Un estudio que siguió a 29 pacientes con SJ que presentaron el signo del molar, con una edad promedio de 8 años 5 meses, reportó que 13 (44,8%) presentaron apnea en el tiempo de seguimiento(13); lo que demuestra la presencia de apneas en la adolescencia y adultez, llevando a complicaciones graves del sueño como en el caso de nuestro paciente y lo descrito en otros reportes(9)(11). Por otro lado, en Argentina se describieron 4 casos de SJ puro en adultos de una misma familia, la madre consultó para conocer la causa de discapacidad intelectual y visual de sus hijos. En la descripción reportaron episodios de apnea hiperpnea que mejoraron progresivamente; no reportan si este síntoma persistió hasta la adultez o si presentaban alteraciones del sueño(2).
En conclusión, son pocos los casos descritos de SJ en la adultez. Es importante tener en cuenta este síndrome para que, en edades gestacionales tempranas, se realice con rigurosidad la ultrasonografía de detalle anatómico para detectar las anormalidades de la fosa posterior que permitan sospechar SJ. De esta manera, se justifica la realización de resonancia magnética nuclear craneal que ofrece más información para establecer el diagnóstico(14). Con el propósito de realizar la detección precoz de posibles alteraciones en otros órganos, es necesario garantizar una atención oportuna por un grupo multidisciplinar de profesionales. La asociación entre SJ y alteraciones del sueño, aunque infrecuente, afecta la calidad de vida del paciente. Por esa razón se hace necesario reunir más información respecto a este síndrome y definir mejor su manejo.