









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCION
Los desórdenes paraneoplásicos producen una diversidad de manifestaciones clínicas por la potencialidad de afectar varias áreas del sistema nervioso central, periférico, unión neuromuscular y músculos, debido a que en su mecanismo de génesis intervienen mecanismos inmunológicos y producción de anticuerpos que afectan sitios distantes y preceden a la detección de los tumores causantes del desorden. Hace 2 décadas apenas se habían identificado 4 entidades de esta naturaleza sin embargo en años siguientes entidades catalogadas como idiopáticas o degenerativas han cambiado en su concepción y así se ha ampliado el espectro de los desórdenes paraneoplásicos gracias a la detección cada vez mayor de autoanticuerpos neuronales (1)(2)(3)(4)(5).
El diagnóstico sindrómico se basa en el fenotipo clínico y en la detección usualmente de uno o más anticuerpos antineuronales con la limitación de que existe superposición de muchos de ellos, mientras que el diagnóstico etiológico se relaciona con el tipo de anticuerpos detectados, lo cual en última instancia define el tipo de tratamiento y la posibilidad de instaurarlo lo más temprano posible y también el pronóstico del paciente. (6)(7).
Nosotros presentamos el análisis de un caso de un Síndrome de Persona Rígida (SPS por sus siglas en inglés) de etiología paraneoplásica que corresponde solo a un 5% de los casos en comparación con la etiología autoinmune que es la más frecuente (8); el diagnóstico sindrómico fue basado en claves clínicas y el diagnóstico etiológico en claves precisas de laboratorio debido a la limitación tecnológica y de presupuesto de nuestra institución para la determinación de un amplio panel de anticuerpos antineuronales.
CASO CLINICO
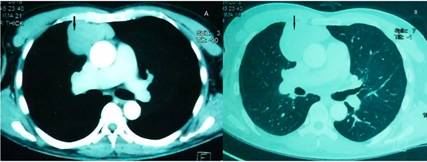
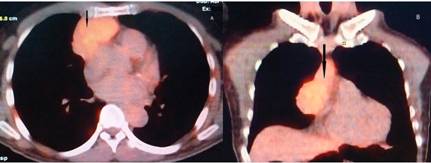
Paciente masculino de 72 años de edad, con historia de hipertensión arterial tratada con olmesartan y amlodipina. Admitido a un primer centro hospitalario por un cuadro de 10 días de evolución de espasmos musculares dolorosos que aparecían en episodios, al inicio en músculos de la región axial tanto dorsal como lumbar y que a los pocos días se extendieron a músculos de miembros inferiores provocando una rigidez axial y apendicular que incrementaba en la noche llevándolo a una postura en hiperextensión axial y de miembros inferiores. Estos episodios, durante el día, eran desencadenados por estímulos táctiles o sonoros. No se definió una etiología y fue medicado con baclofeno y diazepam vía oral con lo que los síntomas mejoraron parcialmente. Sin embargo, en las siguientes semanas el cuadro empeoró: los espasmos se tornaron constantes, lo que le provocó una marcha pseudoespástica y con esto presentó varias caídas y limitación funcional para deambular obligándolo al uso de silla de ruedas. También presentó prurito importante en todo el cuerpo y empezó a tener ansiedad y cambios en su comportamiento con conductas obsesivo-compulsivas. Fue admitido esta vez a nuestro hospital a los 2 meses desde el inicio de los primeros síntomas, encontrando en el examen neurológico rigidez en musculatura paraespinal y en miembros inferiores, hiperekplexia desencadenada por estímulo táctil o sonoro, espasticidad, hiperreflexia en miembros inferiores, clonus aquíleo y una respuesta de triple flexión. Se sospechó un SPS y se inició el trabajo de confirmación sindrómica con una electromiografía que determinó una actividad muscular continua. Se solicitó la determinación de anticuerpos anti GAD 65, anti amfifisina y anti receptores de glycina (ninguno se realiza en nuestro país) de los cuáles solo se pudo realizar el primero. La muestra se remitió a Rochester Mayo Clinic con el siguiente resultado: Ácido Glutámico Decarboxilasa (GAD 65) en líquido cefalorraquídeo (LCR): 43 nmol/L (menor a 0,02) = 4300 U/ml (valor normal 0-5). Un body scan demostró una masa extrapulmonar con inclusión mediastinal anterior de 6x7 cm de diámetro (Figura 1), de densidad homogénea en el rango sólido, un nódulo subpleural en el segmento apical del lóbulo pulmonar inferior derecho de 5mm y otro subpleural posterobasal izquierdo de 3 mm de diámetro respectivamente. Entonces se sometió a tomografía por emisión de positrones (PET CT): F18-FDG: que demostró una masa mediastinal hipermetabólica de 6.6 x 5.5 x 4.7 cm en sus diámetros longitudinal, transversal y anteroposterior (Figura 2). Con estos hallazgos se realizó biopsia percutánea: que determinó mediante el estudio histopatológico: Linfoma No Hodgkin Inmunofenotipo T.
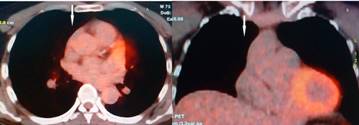
Con todos estos resultados fue confirmado el diagnóstico de SPS de etiología paraneoplásica. Durante su hospitalización recibió: baclofeno, clonazepam, acido valproico, metilprednisolona 1 gr IV diario por 5 días, inmunoglobulina intravenosa (IVIG) a una dosis total de 2 mg/Kg de peso y al confirmar el Linfoma se sometió a quimioterapia con protocolo R-CHOP 6 ciclos y posteriormente radioterapia con 50 Gys. Con este tratamiento se consiguió una recuperación completa con remisión total de los síntomas y reincorporación del paciente a la actividad laboral. Seis meses posteriores se realizó un nuevo estudio de PET CT encontrándose: lesión de tejidos blandos en mediastino anterior, de aspecto residual, con bajo metabolismo glucídico. Criterios de Deauville: 2. Reducción morfometabólica de lesión conocida. Respuesta metabólica completa según criterios Percist (Figura 3).
DISCUSION
El SPS es un desorden neurológico raro con una base inmunológica en el cual anticuerpos ejercen un bloqueo funcional sobre la neurotransmisión inhibitoria sobre el sistema nervioso central (8), este síndrome es más frecuente en mujeres que en hombres y se presenta entre la 4ta y 5ta década de la vida (9)(10)(11).
Nuestro caso es de un paciente de sexo masculino de 72 años que se presentó con rigidez muscular axial y en miembros inferiores, espasmos musculares dolorosos desencadenados por estímulos sonoros y táctiles; compatible con la forma clásica de SPS descrita en la literatura, la misma que inicia con rigidez muscular fluctuante en el tronco sigue a la región lumbar y luego se extiende a músculos de las regiones proximales de las extremidades principalmente piernas, además hiperlordosis lumbar, (3)(7)(12) espasmos musculares dolorosos sobreañadidos catalogados como hiperekplexia adquirida (espasmos precipitados por estímulos externos) (3)(6)(7)(13); en la literatura también se ha descrito una marcha hiper lordótica (13)(14) y pseudo espástica como la reportada en el síndrome de rigidez de piernas (15); este tipo de marcha también presentó nuestro paciente, lo que le ocasionó múltiples caídas. Otros síntomas que se presentan en el síndrome son: alteración del olfato y del gusto, prurito, dificultad en cambios de posición corporal al permanecer acostado (16)(17), síntomas psicológicos como desorden de ansiedad generalizada, depresión, trastorno obsesivo, fobias una de ellas expresada como miedo a caídas (8)(11)(12)(16); estos otros síntomas junto con las caídas actualmente se consideran como complicaciones del SPS (18) y también se encontraron en nuestro paciente.
Los criterios diagnósticos establecidos para esta entidad son:
Criterios mayores: 1. Rigidez muscular en extremidades y musculatura axial en músculos paraespinales tóraco-lumbares que llevan a hiperlordosis, 2. Espasmos episódicos dolorosos sobreañadidos precipitados por estímulos táctiles, ruido inesperado o stress emocional, 3. Electromiografía (EMG): confirmación de actividad continua de la unidad motora en músculos agonistas y antagonistas, 4. Ausencia de otros desórdenes neurológicos o deterioro cognitivo, que pudieran explicar los síntomas de rigidez;
Criterios menores: 1. Presencia de anticuerpos anti GAD 65 o antiamfifisina en suero evaluados por inmunocitoquímica, Western Blot o radioinmunoensayo, 2. Respuesta clínica a benzodiacepinas (19)(20). Todos estos criterios se cumplieron con nuestro paciente.
En el estudio electromiográfico de nuestro paciente se confirmó la presencia de actividad muscular continua en músculos paraespinales, como se encuentra reportado en la literatura, a pesar de alcanzar una relajación muscular durante el estudio, junto a otros parámetros como una relación Hmax/Mmax normal (21)(22).
En los estudios de laboratorio del caso presentado se encontró Ac anti GAD 65 en suero y en LCR. En la literatura se reporta que más del 80% de pacientes con SPS tienen Ac anti GAD 65 en suero o LCR (23)(24), éstos no se correlacionan con la severidad de los síntomas (23) y están asociados frecuentemente con causas autoinmunes (25), sin embargo existen otros anticuerpos detectados, algunos con un rol patogénico potencial como es el caso de Ac Anti GABAA APR, Ac anti gephyrin, Ac anti receptores de glicina, Ac antiamfifisina, éstos 2 últimos asociados con linfoma, cáncer de mama y de pulmón (2)(26)(27)(28). Además, se ha reportado asociación del síndrome con otros tipos de cáncer como: timoma, carcinoma de células renales y adenocarcinoma de colon (2)(6)(7).
Estos anticuerpos producen una respuesta inmuno mediada contra antígenos intracelulares, en el caso del GAD 65, a nivel de las terminales nerviosas presinápticas GABAérgicas disminuyendo la síntesis del GABA (3)(8)(24)(29)(30)(31) induciendo un estado de hiperexcitación neuronal por dos vías: 1. inhibición de las interneuronas a nivel de la médula espinal (vía GABAérgica) y a nivel supraespinal en el tronco cerebral (vía glicinérgica) (14)(20)(25) y 2. por facilitación intracortical a nivel del córtex motor (vía corticoespinal) (32)(33).
Los anticuerpos anti GAD 65 pueden estar presentes en muchos otros síndromes neurológicos (27), pero al constituir un criterio menor de SPS y junto con la clínica el diagnóstico se consideró realizado. No obstante, existen varios fenotipos que se superponen y no necesariamente se correlacionan con un anticuerpo específico por lo que la literatura recomienda realizar todo el panel de anticuerpos para definir la etiología del síndrome. Además, las pruebas paralelas de suero y LCR tienen la mayor sensibilidad y especificidad por lo que constituyen el estándar de oro en el diagnóstico (28) y puesto que en nuestro medio existe una completa limitación de testar todo el panel tanto por lo económico cuanto por lo tecnológico, el reto fue establecer la etiología sin tener la opción de solicitar otro marcador inmunológico (anticuerpo) en suero o en LCR y contando solo con claves clínicas y de laboratorio.
Los grupos etiológicos definidos según el estudio de Sarva H. et al son: autoinmune, paraneoplásico y criptogénico (éste último cuestionado por los propios autores) (16) en donde la edad de presentación fue la clave diagnóstica más importante. La mayor edad media de presentación fue de 59 años en el grupo de etiología paraneoplásica (hasta 81 años) (16); en nuestro caso, la edad es 73 años. La siguiente clave para el diagnóstico fue el tiempo desde el inicio de los síntomas hasta la presentación. El tiempo más corto o evolución más rápida fue de 3 meses en el mismo grupo etiológico (16); en nuestro caso fueron 2 meses.
La siguiente clave diagnóstica fue de laboratorio. En el trabajo de Ariño H et al (34) se analizaron retrospectivamente pacientes con diagnóstico definitivo o posible de síndromes paraneoplásicos sin anticuerpos onconeurales pero en quienes se identificó Ac anti GAD (en suero y en LCR) durante su investigación de etiología paraneoplásica. Se demostró que aquellos que tenían niveles de Ac anti GAD > 2000 U/ml, pacientes de género masculino y de mayor edad (la media de edad en el estudio fue 60 años) tienen un riesgo incrementado de tener un cáncer subyacente, por lo que el trabajo diagnóstico deberá ampliarse para encontrar la etiología. En nuestro paciente, el título de Ac anti GAD fue 4300 U/ml en LCR (valor normal 0-5) lo que motivó a ampliar el trabajo diagnóstico con estudios de imagen como body y pet scan (disponible en nuestra institución) encontrándose una masa mediastinal cuyo resultado histopatológico reveló un Linfoma No Hodgkin Inmunofenotipo T.
En la serie de Sarva H. et al, en el grupo paraneoplásico, el 47% de pacientes tuvieron Ac anti GAD, mientras que la positividad para Ac antiamfifisina fue en el 30% de pacientes. De 19 pacientes en este grupo paraneoplásico, solo 1 (5%) tuvo linfoma de Hodgkin. De los 19 pacientes, 16 (84%) mejoraron con alguna forma de tratamiento oncológico (16).
Nuestro paciente recibió tratamiento sintomático a base de benzodiacepinas asociado a otros GABAérgicos como ácido valproico y baclofeno, todos en vía de administración oral, con lo que tuvo una mejoría parcial. Se inició también inmunoterapia de primera línea a base de metilprednisolona + IVIG por el tiempo de evolución subagudo, tal como lo recomienda en la literatura (12)(18)(28)(35) con lo que mejoró de forma importante. Recibió además tratamiento oncológico con lo que su mejoría fue completa con remisión total de los síntomas a los 6 meses.
CONCLUSION
Este caso es reportado porque al ser el SPS una entidad rara, requiere un alto grado de sospecha etiológica hacia las causas neoplásicas por lo inhabitual que pueden ser (5% de casos) y porque la curación dependerá en gran medida de que el manejo oncológico se instaure en forma temprana. Por tal razón, quizá en nuestro medio, se podría simplificar el diagnóstico y la búsqueda etiológica con títulos de Ac anti GAD 65 > 2000 U/ml que a más de confirmar el diagnóstico clínico brinda altas posibilidades de sospechar un cáncer subyacente como causa del síndrome.