









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La parálisis periódica hipocalémica es un desorden autosómico dominante que se encuentra agrupado entre las canalopatías del músculo esquelético. Se caracteriza por parálisis muscular flácida, causada por disfunción de los canales iónicos de calcio Cav1.1 (por mutación en el gen CACNA1S) o de sodio Nav1.4 (gen SCN4A) localizados en el musculo esquelético (1) . Estas alteraciones conducen a cambios de la permeabilidad de la membrana celular al potasio, disminuyendo sus niveles séricos, y alterando la excitabilidad de la fibra muscular (2) . Las crisis suelen desencadenarse con factores nutricionales, infecciosos, ambientales o con la actividad física. A pesar de que corresponde la forma más común de parálisis periódica es una enfermedad rara, con una prevalencia estimada de 1 caso por 100.000 personas, afectando principalmente pacientes adolescentes, con mayor tasa de crisis de debilidad muscular entre los 15 y 35 años. (3)(4)
La información en nuestro país se limita a 3 reportes encontrados en la literatura; destacando el estudio realizado por Ke Tie y colaboradores en 2009 que identificó una nueva mutación c.2627T>A (p.V876E) localizada en una nueva región de CACNA1S en el segmento S3 del dominio III, como la responsable de la enfermedad en una familia con 6 miembros identificados con la mutación, con una edad temprana (5.2 ± 3.6 años) e inusual de inicio, penetrancia completa y pronóstico severo, al presentarse en dos miembros de la familia muerte temprana por afectación de músculos respiratorios (5)(6).
Se presenta un caso confirmado genéticamente de esta condición y se realiza una revisión de la literatura con énfasis en los aspectos fisiopatológicos y en el impacto de la caracterización de la mutación responsable en el pronóstico y tratamiento de esta enfermedad.
Presentación del caso
Un hombre de 20 años de edad, soldado de las fuerzas militares, asiste a urgencias por 3 días de diarrea acuosa, con frecuencia de 3 deposiciones por día, asociado a debilidad muscular progresiva de las 4 extremidades hasta condicionar limitación completa para la movilidad sin comprometer los músculos de la respiración.
El paciente había consultado en el ultimo año al servicio de urgencias en 5 oportunidades por episodios de debilidad muscular, 4 de ellos posterior a la actividad física, en 2 oportunidades se había documentado la presencia de hipocalemia y los síntomas habían mejorado tras la hidratación y normalización del potasio sérico (Tabla 1). Negaba consumo de sustancias psicoactivas y sus antecedentes familiares eran negativos.
Tabla 1 Paraclínicos de ingreso
| PARACLÍNICOS | RESULTADOS | |
|---|---|---|
| Electrolitos | K: 1.3 mEq/L, Cl: 112 mEq/L, Na 143 mEq/L, Calcio: 8.7 mg/dL | |
| Gases Arteriales | pH: 7.42, pCO2 36.1 mmHg, pO2 86 mmHg, cHCO3 20.1 mEq/L | |
| Otros | CK: 74 umol/L (Normal) |
Al examen físico presentaba frecuencia cardiaca de 62 latidos por minuto, frecuencia respiratoria de 16 respiraciones por minuto, presión arterial 130/72 mmHg y temperatura axilar normal. Su estado de conciencia era normal, sin embargo era llamativa la presencia de hipotonía, arreflexia y debilidad muscular de las 4 extremidades de predominio proximal.
Laboratorios iniciales con hemoglobina de 13,7 gr/dl, leucocitos 9.700 células/ml y plaquetas 175000/ml. Gases arteriales, glucosa, calcio, creatinina, nitrógeno ureico, magnesio, sodio, cloro, glucosa sérica, creatinfosfoquinasa y coproscópico eran normales; destacaban un potasio sérico en 1.3 meq/L y un electrocardiograma anormal con hallazgos relacionados con hipocalemia (Figura 1).
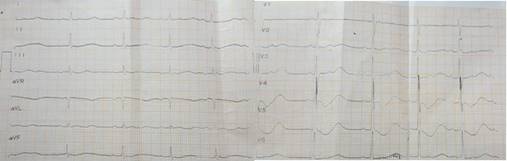
Figura 1 Electocardiograma de ingreso: Ritmo sinusal, FC: 59 lpm, eje: 38° PR: 161 ms, QRS: 94 ms, QTc: 674 ms, infradesnivel del ST en V2 a V5 cóncavo; onda T aplanada y asimétrica, onda U prominente
El paciente ingresó a salas de reanimación para monitorización electrocardiográfica, hidratación y reposición de potasio a través de catéter venoso central; su evolución fue favorable con normalización del potasio sérico, resolución completa de la debilidad y anormalidades electrocardiográficas. Igualmente hubo mejoría de los síntomas gastrointestinales.
Posteriormente es trasladado a sala general de medicina interna; se desarrollaron estudios de potasio en orina de 24 horas, actividad de la renina plasmática, aldosterona sérica, cortisol sérico, TSH, T4 libre, electromiografía y neuroconducciones, los cuales fueron normales (Tabla 2).
Tabla 2 Estudios de extensión y seguimiento para evaluación de hipokalemia
| TIPO DE ESTUDIO | RESULTADOS |
|---|---|
| TSH | 4.27 mUI/L (ref: 0.45 - 4.49 mUI/L) |
| T4L | 1.19 ng/dl (ref: 0.8 - 1.9 ng/dl) |
| Electrolitos | Glicemia basal 79, K: 4.8 mEq/L, Cl: 103 mEq/L, Ca: 9.4 mg/dL, Mg: 1.77 mg/dL |
| Azoados | Creatinina: 0.8 mg/dL, Nitrogeno ureico: 16 mg/dl |
| Potasio en orina | 9 mmol/día |
| Cortisol libre en orina 24 horas | 42.9 ug/24 horas |
| Aldosterona en suero | 53 |
| Renina plasmática | 44.2 |
| Gases arteriales | pH: 7.38, pCO2: 29.6 mmHg, cHCO3: 19.8 mEq/L, paO2: 62 mmHg |
| Glicemia pre y postcarga de glucosa | Precarga: 72 mg/dL; poscarga: 81.1 mg/dL |
| Hb1Ac | 5,1% |
| Ecocardiograma transtorácico | Buena función sistólica y diastólica ventricular FEVI 65%, sin lesión valvular |
| Ecografía renal y vías urinarias | Estudio sin alteraciones ecográficas |
| Electromiografía y neuroconducción | Estudio neurofisiológico de 4 extremidades con potenciales de nervios motores y sensitivos con latencias, amplitud y velocidades normales. Electromiografía con silencio eléctrico en reposo, actividad de inserción normal, reclutamiento y activación adecuados y PUM* de morfología normal |
*PUM: Potenciales de Unidad Motora
La historia de episodios de parálisis periódica desencadenados por el ejercicio y un episodio actual relacionado con gastroenteritis aguda viral asociados a hipocalemia con excreción renal normal de potasio y la exclusión de otras causas de hipocalemia, nos orientó al diagnóstico de parálisis periódica hipocalémica primaria (Tabla 3). Se realizó test de provocación con ejercicio el cual fue negativo. En conjunto con servicio de genética clínica se solicitó secuenciación para genes CaCNa1s y SCN4a; identificándose variante patogénica CACNA1S c.1583G>A;p.(Arg528His).
Tabla 3 Criterios diagnósticos para Parálisis Periódica Hipocalémica
| Criterios diagnósticos de apoyo para parálisis periódica hipocalémica |
| 1. Dos o más ataques de debilidad muscular con potasio sérico documentado <3,5 mEq/L |
| 2. Un ataque de debilidad muscular en el probando y un ataque de debilidad en 1 familiar con K+ sérico documentado <3.5 mEq/L en al menos 1 ataque |
| 3. Tres de las 6 características clínicas o de laboratorio: a. Inicio en la primera o segunda década. b. Duración del ataque (debilidad muscular que involucra 1 o más extremidades)> 2 horas c. Desencadenantes positivos (comida rica en carbohidratos, descanso después del ejercicio, estrés) d. Mejora con la ingesta de potasio e. Antecedentes familiares positivos o mutación esquelética genéticamente confirmada del calcio o del canal de sodio f. Prueba de ejercicio largo positiva de McManis |
| 4. Exclusión de otras causas de hipokalemia (disfunción renal, suprarrenal, tiroides, acidosis tubular renal, abuso de diuréticos y laxantes) |
| 5. Ausencia de miotonía (clínicamente o latente detectada por la aguja EMG), excepto los párpados |
*Tomado de: Jeffrey M. Statland MD Bertrand Fontaine MD, PhD Michael G. Hanna MD Nicholas E. Johnson MD John T. Kissel MD Valeria A. Sansone MD . Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle & Nerve 2017 Volume 57, Issue 4.
Basados en la alteración genética encontrada se decidió inicio de tratamiento con acetazolamida a dosis de 250 mg cada 12 h con buena tolerancia. Se dieron recomendaciones generales con énfasis en las medidas abortivas de crisis de debilidad, consejería nutricional, genética, y seguimiento multidisciplinario ambulatorio presentando adecuado control de los síntomas sin nuevos episodios de debilidad documentados.
Discusión
El potencial de acción juega un papel esencial de los fenómenos de contracción- relajación de la fibra muscular y es responsable del movimiento del aparato locomotor. De las mutaciones de los canales sarcolémicos reconocidas como responsables de la parálisis hipocalémica episódica, aquella que codifica el receptor de dihidropiridina Cav1.1 del canal de calcio tipo L (CACNA1S) es la más frecuente (80% de los casos), como fue el caso de nuestro paciente.(7) Se han identificado tres tipos de mutaciones sin sentido en el gen CACNA1S más comunes, como resultado de sustituciones en un solo aminoácido, de arginina por histidina o por glicina: en el segmento S4 del dominio II (Arg528His) de la subunidad alfa del canal de calcio; S4 del dominio IV (Arg1239His); y Arg1239Gly. Esta mutación se ubica en el sensor de voltaje de la célula muscular interrumpiendo su función, pues neutraliza los residuos de arginina cargados positivamente en uno de los segmentos (S4) del canal. La resultante es la formación de una corriente de poro de apertura no selectiva a través de dicho sensor cuando éste se encuentra en estado de reposo, pero no en el estado activo, con aumento de la fuga hacia el interior de corrientes de sodio generando una sobrecarga intracelular de este ion y conduciendo a una despolarización sostenida de la fibra muscular que se traduce clínicamente en parálisis flácida de los músculos esqueléticos.(8)(9)
El potencial de membrana en reposo es controlado principalmente por la permeabilidad de la membrana celular al potasio y su conservación es fundamental en diversos procesos fisiológicos esenciales de la vida del ser humano como la contracción muscular.(10) En la parálisis hipocalémica episódica, la disfunción en el sensor de voltaje de los canales Cav1.1 o Nav1.4 produce corrientes de poro de activación protónica que generan un cambio en el pH intracelular hacia un ambiente ácido, inhibiendo el componente externo (R1 y R2) de las corrientes rectificadoras de potasio (iKr) y estableciendo una corriente interna persistente de cationes que hiperpolariza la membrana y despolariza el sarcolema, conduciendo a una despolarización celular paradójica con un potasio extracelular bajo característica de esta condición.(11)(12)(13)
Por su baja prevalencia y la carencia usual de estudios de confirmación genética en nuestro medio, fue necesario en nuestro caso clínico realizar un abordaje diagnóstico completo de la hipocalemia apoyados en un grupo multidisciplinario y priorizando inicialmente la corrección del trastorno hidroelectrolítico severo que podía conducir a desenlaces fatales. Se excluyeron trastornos tiroideos y otras endocrinopatías como hipercortisolismo o hiperaldosteronismo, los gases arteriales y el estudio de electrolitos en orina no fueron sugestivos de tubulopatía y el ecocardiograma transtorácico no reveló ninguna alteración estructural cardiaca asociada. A pesar de que las pruebas de electro-diagnóstico fueron negativas, se consideró que la sospecha de parálisis hipocalémica periódica era lo suficientemente alta para solicitar el estudio genético que finalmente confirmó el diagnóstico como ocurre en el 60-70% de los casos. (14)
La ausencia de un antecedente familiar de esta enfermedad en nuestro paciente puede ser explicado por 2 hipótesis principales. La primera es que, aunque se reconoce que esta enfermedad tiene un patrón de herencia autosómico dominante se han reportado casos esporádicos asociados a otras mutaciones que tienen penetrancia y pronóstico variable como son la mutación ubicada en el segmento transmembrana S3 con inicio de los síntomas a edad más temprana y con un pronóstico severo o las mutaciones R528H y R1239H que tienen cursos clínicos diversos.(14) Otra alternativa, es que la penetrancia variable de la enfermedad condicionada por diversos factores genéticos permite la aparición de manifestaciones clínicas en pocas personas de un grupo familiar. Esto fue demostrado al realizar pruebas de secuenciación genética a los parientes maternos de una paciente femenina con parálisis periódica hipocalémica en Japón, donde se demostró que su madre y abuela eran portadoras de una mutación sin sentido reconocida como causante de la enfermedad, pero carecían de expresión clínica.(15)(16) Por esta razón, el paciente se encuentra en seguimiento por el servicio de genética para determinar si se trata de una mutación de novo o es hijo de un padre portador asintomático para lo cual requiere pruebas genéticas complementarias y la construcción de un pedigree con el fin de aclarar el tipo de herencia de dicha mutación.
La mutación encontrada en nuestro paciente: sustitución c.1583G>A (p.Arg528His), se ha asociado a mayor susceptibilidad de hipertermia maligna tipo 5 que es una afección farmacogenética que conduce a un estado hipermetabólico del músculo esquelético que es potencialmente mortal, inducido por la exposición a ciertos medicamentos (anestésicos volátiles o halogenados y/o bloqueadores neuromusculares despolarizantes como la succinilcolina) o ante situaciones estresantes como ejercicio extenuante o calor excesivo y que se caracteriza por producir hipertermia y falla multiorgánica secundario a rabdomiolisis. Esto se explica por la asociación de la mutación en el gen del receptor 1 de rianodina (RYR1), y la mutación CACNA1S que se presenta en el 1% de los casos, pero puede conducir a hipertermia maligna casi siempre fatal en ausencia de un tratamiento oportuno.(17)(18) Por esta razón, se indicó al paciente la necesidad de tener una reubicación laboral en las fuerzas militares y que en caso de requerir un procedimiento quirúrgico se debe aclarar esta asociación en la valoración pre-anestésica para vigilar reacciones similares a la hipertermia maligna y evitar complicaciones.
Las alternativas de tratamiento farmacológico de esta enfermedad son limitadas (inhibidores de anhidrasa carbónica y antagonistas del receptor de aldosterona), siendo el pilar del tratamiento las medidas no farmacológicas que permitan prevenir o abortar los episodios de crisis de debilidad.(19) La importancia de la caracterización genética de la enfermedad radica en que los efectos nocivos de las sustituciones de histidina podrían mejorarse con un pH más ácido, mientras que las mutaciones de glicina son insensibles a las alteraciones del pH que producen los inhibidores de la anhidrasa carbónica. (20)Por esta razón, se ha observado que aquellos pacientes con la mutación SCN4A que contengan sustitución de glicina no se benefician de la acidosis metabólica que genera la acetazolamida.(21)(22) Por el contrario, se han reportado variantes fenotípicas asociadas a mutaciones puntuales sin sentido que manifiestan una respuesta negativa al manejo con acetazolamida y una mejoría marcada de síntomas de debilidad muscular con una combinación de espironolactona, amilorida y suplementos de potasio.(23)(24)
Conclusiones
La parálisis hipocalémica periódica es una condición clínica poco prevalente que se agrupa dentro de las canalopatías del músculo esquelético y se caracteriza por episodios de parálisis muscular flácida por alteración de los mecanismos de excitación - contracción muscular. La importancia de nuestro reporte radica en el abordaje diagnóstico realizado para descartar otras causas potenciales de hipocalemia y la confirmación genética de la mutación responsable. Se destaca la relevancia de la caracterización genética en esta entidad dado su impacto en las intervenciones terapéuticas y la probabilidad de responder a las medidas farmacológicas y de esta manera controlar la recurrencia de los episodios de debilidad muscular propios de esta entidad