









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La AP (OMIM # 606054) es una de las AO más comunes, con una incidencia aproximada de 1 en 100,000 a 150,000 RN (1). Esta entidad es causada por la deficiencia de propionil-CoA carboxilasa (PCC), enzima mitocondrial que cataliza la conversión de propionil-CoA en metilmalonil-CoA utilizando biotina como cofactor (2)(3). La enzima está compuesta de 2 subunidades α y β codificadas por sus respectivos genes, PCCA y PCCB sin presentar una clara correlación genotipo-fenotipo (4)(5)(6). El bloqueo metabólico da como resultado el acúmulo de ácido propiónico y metabolitos relacionados con propionil-CoA, que pueden detectarse bioquímicamente, constituyendo sellos bioquímicos para el diagnóstico. La acumulación de estos metabolitos afecta el adecuado funcionamiento de otras vías metabólicas como el ciclo de la urea, el metabolismo de la glicina y la gluconeogénesis ocasionando mayor toxicidad y disfunción en el sistema nervioso central y cambios en el metabolismo de los aminoácidosc (7)(8)(9)(10).
La AP fue la primera AO descrita en la década de 1960 (11), definiendo un defecto generalizado que producía pérdida del grupo amino de ciertos aminoácidos en el músculo, ocasionando hiperamonemia y cetoacidosis, al igual que exceso de glicina en fluidos biológicos, por lo que también se le conoce como hiperglicinemia cetósica (4)(12).
La AP presenta alta tasa de mortalidad con manifestaciones clínicas heterogéneas (6).
Las características clínicas del espectro AP varía desde el inicio neonatal hasta la enfermedad de inicio tardío (13):
1. AP de inicio neonatal es la forma más común (80% de los casos), se caracteriza por presentarse en el RN después de un corto periodo libre de síntomas, alimentación deficiente con rechazo y vómitos, disminución del estado de conciencia, seguida de encefalopatía progresiva de origen inexplicado, precipitados por la ingesta de proteínas y de varios aminoácidos, principalmente leucina. Sin un diagnóstico y manejo rápidos, la sintomatología progresa con letargo, convulsiones o coma que puede causar la muerte. Las alteraciones bioquímicas más frecuentemente asociadas son: acidosis metabólica con anion gap elevado, acidosis láctica, cetonuria, hiperglicinemia, hiperglicinuria, hiperamonemia, citopenias e identificación de 3-hidroxi-propionico, propionilglicina y metilcitrato (6)(14)(15).
2. La AP de inicio tardío intermitente, pueden permanecer asintomáticas y sufrir crisis metabólica bajo estrés catabólico (ej: infecciones, enfermedad, cirugía o ayuno) presentando letargia, ataxia, anorexia, vómitos, retraso del desarrollo y recurrentes estados de coma cetoacidótico. Se acompaña de hiperamonemia, hiperglicininemia y citopenias, en especial neutropenia (1).
3. La presentación tardía crónica progresiva de AP cursa con inicio insidioso con el desarrollo de complicaciones multiorgánicas que incluyen vómitos, intolerancia a proteínas, neuropatía óptica, cardiomiopatía tanto dilatada como hipertrófica y arritmias, nefromegalia (16)(17)(18)(19), osteoporosis, retraso del crecimiento, hipotonía, déficits neurocognitivos, regresión del desarrollo, psicosis, trastornos del movimiento (distonías, o coreoatetosis) (9)(20).
Los progresos realizados en el tratamiento han mejorado la supervivencia de estos pacientes así como la frecuencia de crisis; sin embargo no han evitado el deterioro cognitivo o complicaciones a largo plazo progresivos con insuficiencia de órganos selectivos (21)(22).
CASO CLÍNICO
La AP presenta alta tasa de mortalidad con manifestaciones clínicas heterogéneas (6). Reportamos el caso de una RN femenina, que ingresa a los 7 días de vida, producto de tercera gestación de madre de 29 años, segundo embarazo con actual pareja y antecedente de un hijo fallecido antes de los 3 meses por trastorno metabólico no especificado, otro hijo de 9 años sano de otro padre. G3P3V2M1, refiriendo adecuado control prenatal. Nace a las 38 semanas por parto normal, adecuada adaptación neonatal y egreso hospitalario. Al tercer día de vida presenta hipoactividad, hiporexia y letargo, por lo que es llevado al hospital local, se documentan glicemia de 50 mg/dl e inician líquidos endovenosos. El paciente presenta posterior parada cardiaca de corta duración que requiere reanimación, la cual fue exitosa. Al sexto día de vida presenta deterioro clínico dado por dificultad respiratoria, cianosis, bradicardia por lo que deciden iniciar ventilación mecánica y manejo antibiótico de primera línea, posteriormente se evidencia acidosis metabólica, se inicia bicarbonato de sodio y soporte inotrópico, remitiendo a institución de mayor complejidad, en donde ingresa en condiciones clínicas críticas, bradicárdica, hipoglicemia, requiere reanimación posterior a su ingreso, bolos de cristaloides endovenosos, bicarbonato, múltiple soporte inotrópico, corrigiendo hipoglicemia. Requiere soporte con ventilación mecánica convencional, posteriormente ventilación mecánica de alta frecuencia, se intenta corregir desequilibrio electrolítico con líquidos endovenosos, sin recibir aporte nutricional por sus condiciones clínicas. La paciente realiza varios paros cardiacos con estado de choque refractario a pesar del manejo, la paciente fallece a los 8 días de vida. La muerte ocurrió antes de que se tuviera el diagnóstico bioquímico.
El reporte de laboratorio informó hemograma: Leucocitos: 2.950 Neutrófilos: 27% Linfocitos: 69% Hemoglobina: 11. 2 gr/dl Hematocrito: 33.3% Plaquetas: 108.000. Bilirrubina total 7. 01 mg/dl Bilirrubina Indirecta 5. 6 mg/dl Glicemia 47 mg/dl AST 345. 3 UI/L. ALT: 275. 2 UI/L Proteínas totales: 3.45 gr/L Albumina 2. 46 gr/L Amonio 117.4 μmol/L (VN: 64-107 μmol/L). Gases arteriales: PH 7. 26 PCO2:34 PO2:33 HCO3:16. 6 Acidosis metabólica con hipoxemia. Se toman muestras para análisis de ácidos orgánicos en orina y cuantificación de aminoácidos en plasma por cromatografía.
Se identificó leve hiperglicinemia: 1.053 mmol/mol de creatinina (VR: 179 - 941) y cistinemia: 30 mmol/mol de creatinina (VR: 3-17), asociados a hiperlisininemia severa: 1.471 mmol/mol de creatinina (VR: 16 - 173) en la cromatografía por HPLC de aminoácidos en plasma (Tabla 1).
Tabla 1 Resultado de aminoácidos cuantitativos en plasma por HPLC
| NOMBRE DEL ESTUDIO | VALOR PACIENTE (µmol/L) | VALOR REFERENCIA (µmol/L) |
| ASPARAGINA | 80 | 32 - 98 |
| ACIDO GLUTAMICO | 22 | 34 - 177 |
| GLUTAMINA | 520 | 204 - 1101 |
| SARCOSINA | 0 | 0 - 145 |
| GLICINA | 636 | 94 - 553 |
| ALANINA | 520 | 161 - 535 |
| CITRULINA | 4 | 1 - 33 |
| ACIDO ALFA AMINOBUTIRICO | 26 | 2 - 51 |
| VALINA | 193 | 84 - 318 |
| CISTINA | 10 | 6 - 43 |
| METIONINA | 15 | 10 - 45 |
| CISTATIONINA | 3 | 0 - 3 |
| ISOLEUCINA | 76 | 21 - 90 |
| LEUCINA | 162 | 58 - 171 |
| LISINA | 714 | 35 - 220 |
| TIROSINA | 120 | 39 - 86 |
| BETALANINA | 0 | 0 - 64 |
| FENILALANINA | 19 | 37 - 62 |
| BETAMINO ISOBUTIRICO | 0 | 0 - 99 |
| HOMOCISTEINA | 0 | 0 - 1 |
| AC. GAMA AMINOBUTIRICO | 3 | 0 - 3 |
| ETANOLAMINA | 49 | 0 - 42 |
| HIDROXILISINA | 0 | 0 - 22 |
| ORNITINA | 32 | 16 - 129 |
| 1- METILHISTIDINA | 6 | 2 - 19 |
| HISTIDINA | 28 | 47 - 95 |
| TRIPTOFANO | 7 | 47 - 95 |
| 3 -METIL HISTIDINA | 20 | 0 - 4 |
| ANSERINA | 0 | 0 - 11 |
| CARNOSINA | 0 | 0 - 10 |
| ARGININA | 5 | 13 - 128 |
| PROLINA | 237 | 80 - 664 |
| ACIDO ASPÁRTICO | 8 | 0 - 31 |
| TREONINA | 61 | 45 - 194 |
| SERINA | 148 | 73 - 154 |
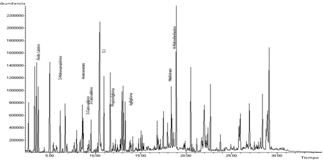
La cromatografía de ácidos orgánicos en orina identificó los ácidos: 3-hidroxi-propiónico, metilcitrato, propionilglicina, 2-metil-3-hidroxibutírico, metilcítrico, 3-hidroxibutírico, 4 hidroxifenilláctico, ácido láctico, ácido acético, Ácido glutárico, 2-hidroxiisovalérico, 2-hidroxiisobutírico (Figura 1). La presencia de los tres primeros ácidos orgánicos confirma el diagnóstico de AP.
DISCUSIÓN
La AP es un error innato del metabolismo, autosómico recesivo que pertenece al grupo llamado acidemias o acidurias orgánicas, caracterizándose por la excreción de ácidos orgánicos en la orina (7)(15). En su afectación multisistémica es fundamental la identificación temprana, especialmente de la forma neonatal de la AP que se caracteriza por cetoacidosis, rechazo al alimento, letargia, retraso del crecimiento y encefalopatía, para realizar una intervención médica oportuna y reducir su alta tasa de mortalidad (13)(20)(23).
En nuestra paciente se evidencian de forma temprana los síntomas clínicos de rechazo de la alimentación, letargia e hipoactividad, con evidencia en laboratorios de leucopenia y trombocitopenia leve, acidosis metabólica e hipoglicemia, como se describe en la forma de presentación neonatal.
Además, tenía el antecedente del fallecimiento de un hermano en edad lactante menor, que presentó sintomatología similar sin llegarse a establecer el diagnóstico, lo que debe alertar al personal médico sobre la posibilidad de estar ante un error innato del metabolismo.
La miocardiopatía y las afectaciones neurológicas se asocian con una alta tasa de morbi-mortalidad y se han atribuido a fallas bioenergéticas y acumulación intracelular de metabolitos tóxicos (24). Como en el presente caso, que a pesar del manejo agudo del desequilibrio ácido base terminó falleciendo.
Las pruebas de ácidos orgánicos en orina en personas que son sintomáticas o aquellas detectadas por el examen de detección en el RN revelan una elevación del 3-hidroxipropionato y presencia de metilcitrato, tigliclglicina y propionilglicina, que establecen el diagnóstico bioquímico como ocurrió en nuestra paciente se reportaron.
La primera alteración observada del metabolismo de los aminoácidos fue la hiperglicinemia, que se detecta en casi todos los pacientes con AP. La hiperlisininemia se sugiere que puede presentarse por la inhibición de la sacaropina deshidrogenasa por el propionil-CoA o sus metabolitos, además asociándose a hiperamonemia (7).
En el presente caso se destaca la utilidad de identificar cistinemia y sobretodo hiperlisininemia, que aunque se han asociado a la patología, no son un hallazgo común y en muchos casos se desconocen como indicativos de AP. La alteración de estos aminoácidos incrementa la acumulación de productos altamente tóxicos que de por si causa la disfunción de la enzima mitocondrial PCC. Por esta razón, la calidad de vida de los pacientes de AP está profundamente afectada cuando se logra sobrevivir (21).
El caso presentado constituye un buen ejemplo de lo diversas que pueden ser las consecuencias metabólicas de la deficiencia de la PCC y la posibilidad de encontrar indicios de esta patología tanto en los paraclínicos iniciales, así como en exámenes especializados como los aminoácidos en plasma, los cuales aunque no constituyen el examen confirmatorio, permiten direccionar la sospecha diagnóstica.
CONCLUSIONES
Se describió un caso clínico de inicio neonatal con reporte confirmatorio de ácidos orgánicos en orina para AP y se asoció de hiperglicininemia e hiperlisininemia severa, que colaboró con deterioro multisistémico conllevando a la muerte de la paciente en la primera semana de vida.
A pesar de la baja incidencia de la AP, ésta debe ser un diagnóstico diferencial a tener en cuenta en especial en la edad neonatal cuando se presenta una sintomatología clínica compatible, acidosis metabólica, leucopenia, hipoglicemia sin etiología esclarecida y determinarse las concentraciones de aminoácidos para el tratamiento de la AP, idealmente en condiciones óptimas, en el menor tiempo posible cuando existan factores de riesgo y realizar ajustes en la terapia nutricional reduciendo precursores de propionil-CoA que impactarían en el pronóstico de los pacientes afectados (25)(26)(27).