









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad de Huntington (EH) es una enfermedad neurodegenerativa rara con herencia autosómica dominante, cuenta con una prevalencia de 5-10 personas afectadas por 100,000 en la población caucásica(1). Se estima que en el 2017 existían 8,000 pacientes con enfermedad de Huntington en México(2). Clínicamente la enfermedad se caracteriza por síntomas motores, psiquiátricos y cognitivos. La EH es causada por una repetición anormal del triplete CAG que codifica para el aminoácido glutamina. El número normal de repetidos es de hasta 26 repeticiones; un número de repetidos mayor o igual a 27 se considera anormal. Lo anterior conlleva a una sobreexpresión de glutamina la cual conlleva a una alteración de la proteína huntingtina ubicada en el cromosoma 4p. La presencia de más de 40 repetidos CAG establece el diagnóstico de EH definitiva.
La EH se puede presentar en cualquier momento de la vida, sin embargo, esta patología usualmente se manifiesta en la edad adulta (30 a 40 años de edad). Se ha establecido que menos del 10% de estos enfermos presentarán la enfermedad antes de los 21 años de edad, a estos se le conoce como enfermedad de Huntington juvenil (EHJ) (4)(5). Por otra parte, si los síntomas se presentan antes de los 10 años de edad se denomina de inicio infantil (4). La mayoría de los casos con EHJ manifestarán una clínica caracterizada por cuadro de rigidez y acinesia conocido como variante Westphal, esto a diferencia de los pacientes con edad de inicio habitual quienes cursan predominantemente con corea.
Las publicaciones de casos juveniles e infantiles, es escasa en la población mundial, y México no es la excepción. Los estudios publicados en población mexicana disponibles a la fecha no están dirigidos a determinar la prevalencia y/o clínica de los casos juveniles (6)(7)(8)(9). El propósito de este estudio es conocer la prevalencia de casos juveniles en una muestra de sujetos con EH confirmada y presentar sus principales características clínicas.
Método
Se trata de un estudio transversal, en el cual se incluyeron pacientes con diagnóstico molecular de EH que acudieron a la clínica de trastornos del movimiento del Instituto Nacional de Neurología y Neurocirugía.
Se revisaron un total de 198 registros de pacientes con EH atendidos entre enero de 2011 y diciembre de 2018. Se identificó a aquellos con inicio de síntomas motores antes de los 21 años de edad. Se llevó a cabo la recolección de los siguientes datos: antecedentes heredofamiliares, personales patológicos y no patológicos, así como historial de la EH incluyendo edad, género, síntomas motores (distonía, rigidez, acinesia, y mioclonías) y tratamiento. Asimismo, se recolectó la presencia y frecuencia de severidad cognitiva, y síntomas psiquiátricos, estos últimos como depresión, ideación suicida, ansiedad, agresividad, y psicosis.
Adicionalmente se recolectó la puntuación de la Escala Unificada de la Enfermedad de Huntington: parte motora (UHDRS-motor), número de repetidos en el alelo mutado, y hallazgos de resonancia magnética. De estos instrumentos mencionados tenemos el UHDRS-motor donde se examinan aspectos motores, este contiene 31 ítems, con una escala del 0 al 4: 0 normal (sin anormalidades motoras); 1 anormalidades motoras no especificas; 2 anormalidades motoras que pueden ser signos de EH (50-89% de confianza); 3 anormalidades motoras que pueden ser signos probables de EH. (90-98% de confianza); 4 anormalidades motoras que son signos inequívocos de EH, con base a lo anterior el puntaje máximo para esta prueba es de 124 puntos. Los estudios de resonancia magnética (RM) de encéfalo (T1, T2 Flair) se realizaron en un equipo de 1.5 teslas.
El estudio cuenta con la aprobación del Comité de Ética local y todos los participantes, o en su caso los tutores legales, otorgaron su consentimiento informado por escrito.
Resultados
Del total de 198 pacientes con EH, el 6.5% correspondieron a EHJ.
Características demográficas
Se incluyeron 12 pacientes con inicio juvenil, y un paciente de inicio en la niñez (8 años). Seis (46.2%) de ellos correspondieron al género femenino, y siete (53.8%) al masculino. El rango de edad de inicio de 8 a 21 años, con una media de 17.8 ± 3.9 y (mediana de 20). El tiempo medio de evolución de la enfermedad fue de 12 ± 3.9 años.
La manifestación clínica inicial fue sintomatología psiquiátrica en el 53.8%; el 38.4% inicio con sintomatología motora y solo un 7.6% (n=1) con alteraciones cognitivas.
Características genéticas
Respecto a herencia; el 69.2% (n= 9) presentó herencia paterna y el 30.7% por herencia materna (n=4).
El rango de número de repetidos de CAG fue de 52 a 73, con una media de 59.9 ± 6.7.
Características clínicas
Los datos demográficos, clínicos y genéticos de los sujetos afectados se resumen en la Tabla 1.
El 53.8% (n=7) presentó corea como síntoma motor predominante; por otra parte, el 38.4% (n=5) manifestó cuadro rígido acinético. Se reportaron otros síntomas motores durante la progresión tales como distonía encontrado en un 38.4% (n=5), temblor en el 23% (n=3), y un paciente con mioclonías. La puntuación del UHDRS-motor con un puntaje mínimo de 28, y máximo de 83 puntos, con una media de 46.2 ± 17.4, y mediana de 40.
Tabla 1 Datos demográficos, clínicos y genéticos de los casos de enfermedad de Huntington juvenil.
| # | Género | Transmisión | Edad inicio de síntomas | Tipo de inicio de síntomas | Síntoma de Inicio de cuadro motor | UHDRS 1 | Años de evolución | Repetidos CAG 5 |
|---|---|---|---|---|---|---|---|---|
| 1 | M2 | Madre | 20 | Psiquiátrico | Corea | 47 | 10 | 24/52 |
| 2 | F3 | Padre | 21 | Psiquiátrico | Corea | 40 | 8 | 17/52 |
| 3 | M | Madre | 20 | Motor | Corea | 35 | 9 | 19/62 |
| 4 | M | Padre | 8 | cognitivo | Westphal | 28 | 21 | 19/73 |
| 5 | F | Madre | 20 | Motor | Corea | 32 | 10 | 25/56 |
| 6 | M | Padre | 15 | Psiquiátrico | Westphal | ND4 | 20 | 17/61 |
| 7 | M | Padre | 20 | Psiquiátrico | Distonía | 83 | 10 | 29/67 |
| 8 | F | Padre | 21 | Psiquiátrico | Corea | 31 | 12 | 18/54 |
| 9 | F | Padre | 21 | Motor | Corea | 66 | 9 | 16/60 |
| 10 | F | Padre | 16 | Motor | Westphal | ND4 | 9 | 18/53 |
| 11 | F | Padre | 12 | Psiquiátrico | Westphal | ND4 | 12 | 29/70 |
| 12 | M | Padre | 21 | Psiquiátrico | Corea | ND4 | 14 | 19/55 |
| 13 | M | Madre | 17 | Motor | Westphal | 54 | 12 | 18/64 |
1 escala unificada para la enfermedad de Huntington (UHDRS). 2 masculino (M). 3 femenino (F). 4 no disponible (ND). 5 tripletes de aminoácidos: citosina, adenina, guanina (CAG).
Alteraciones neuropsiquiátricas
El 84.6% de los casos presentaron alguna alteración neuropsiquiátrica. La afección psiquiátrica más prevalente fue depresión (69.2%) destacando ideación o intento de suicidio en el 30.7% de los mismos. Otras alteraciones reportadas fueron agresividad en el 23%, ansiedad en un 15.3%, y psicosis en el 7.6%.
Deterioro cognitivo
Todos los pacientes presentaron algún grado de deterioro cognitivo, se encontró deterioro cognitivo leve en el 9% de los casos, moderado en el 63.6%, y severo en el 27.2%. En todos los casos se reportaron alteraciones en funciones ejecutivas de moderadas a severas; mientras que en solo dos pacientes se registró afección en procesos práxicos y gnósicos.
Estudios de Imagen
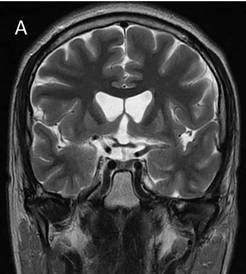
El estudio de neuroimagen reportó mayor frecuencia de atrofia severa con 38.4%, en un 23% moderada, y en 23% leve. En 15.3% de casos se reporta imagen sin alteraciones. En la Figura 1 se muestra estudio de imagen de paciente que corresponde al caso infantil.
Terapéutica
El tratamiento recibido para las alteraciones motoras fue olanzapina en el 30.7% (n=4), amantadina en el 23% (n= 3), haloperidol en el 23% (n=3), clonazepam en el 23% (n=3), biperideno en el 7.6% (n=1), pramipexol en el 7.6% (n=1) y, levodopa en el 15.4% (n=2).
Con relación al tratamiento indicado para síntomas psiquiátricos, el 30.7% (n=4) de los pacientes recibió tratamiento con fluoxetina, el 30.7% (n=4) con valproato de magnesio, 7.6% (n=1) recibió escitalopram, y el 7.6% mirtazapina.
Discusión
Fue en 1872 cuando George Huntington describe por primera vez a la enfermedad de Huntington, describiendo sobre la edad de presentación que “la tercera peculiaridad de la enfermedad es su aparición, al menos como una enfermedad grave, solo en la vida adulta. No conozco un solo caso que haya mostrado signos marcados de corea antes de los treinta o cuarenta años “(10).
En retrospectiva, la publicación inicial de estos casos singulares de EH comenzó como reporte de casos, pero en la actualidad se ha reconocido su relevancia y han tomado lugar las publicaciones descriptivas en poblaciones específicas (11).
No existen a la fecha descripciones clínicas de pacientes mexicanos con EHJ. Alonso y colaboradores (6) publicaron sobre las características genéticas de casos con EH en México; de la muestra recabada se obtuvo que el 8.5% correspondían a casos con EHJ. Sin embargo, dicho estudio no presentó datos clínicos motores, psiquiátricos o cognitivos de la muestra.
La literatura internacional reporta que al menos el diez por ciento de las personas con la EH desarrollan síntomas antes de los 21 años y más del 90% de los mismos ha heredado la enfermedad por vía paterna (12)(13)(14). En el presente estudio el 6.5% de casos correspondió a EHJ y la mayoría de ellos con herencia paterna. Para el caso de Huntington infantil, a nivel mundial se ha reportado una incidencia de entre el 0.5% a 2% de los pacientes con EH (15)(16)(17). En el presente estudio se tuvo únicamente un paciente con inicio infantil (0.5%).
En Grecia, (18), Panas y cols estudiaron un total de 248 casos con EH, de estos siete fueron casos juveniles en los que se reportaron edad y repetidos CAG. La edad media de inicio de síntomas fue de 16.6 ± 1.9 años y la media de repetidos CAG de 55.6 ± 5.6 (rango 48-62). Lo cual es similar a nuestra población de estudio.
En concordancia con los hallazgos encontrados por otros autores (19)(20)(21)(22). Los síntomas más frecuentes de aparición de la enfermedad incluyen alteraciones cognitivas y del comportamiento.
En el estudio aquí presentado la manifestación motora dominante fue corea en la mayoría de los casos (53.8%). Contrario a lo documentado en la literatura donde la sintomatología motora de los pacientes con EHJ es muy diferente al de inicio adulto, este cuadro de aparición temprana en particular incluye rigidez, bradicinesia, distonía, parkinsonismo e incluso epilepsia de difícil control, y es poco común encontrar corea (23)(24). Un reporte realizado en población de los Países Bajos (25) donde se reportaron 53 casos juveniles, corroboró el predominio de clínica de rigidez sobre la corea (60.3% versus 39.6%). Otros estudios como el realizado en Argentina (26), con una descripción de 14 casos, reportaron que el 85.7% de los sujetos con EHJ mostraron en tan solo el 25% (n=3) predominó la corea como manifestación dominante en el cuadro clínico motor de estos pacientes. El sustrato patológico del porqué este cuadro rígido-acinético predomina sobre la corea ha sido atribuido al daño severo en el globo pálido o áreas periventriculares del núcleo caudado (27) en comparación a otras estructuras. Sin embargo, no se excluye la posibilidad de daño en otros sitios con variaciones en los niveles de neurotransmisores que conduzcan a la predominancia de corea u otro síntoma motor sobre la variante Westphal. Otra posible explicación de la predominancia de un fenotipo no habitual en los pacientes aquí reportados, pudiera ser la edad, ya que los pacientes que predominaron con corea iniciaron a los 20 y 21 años de edad, a diferencia de los casos más tempranos en quienes predominó la variante Westphal.
Por otra parte, se ha reportado que la demencia es más prominente en este grupo de edad y presenta una rápida progresión que resulta en un deterioro mental temprano y severo (28). Todos los pacientes de la muestra presentaron algún grado de deterioro cognitivo desde leve hasta demencia subcortical, esto siendo consistente con lo reportado en otras poblaciones (19)(20). Se requieren estudios de cohorte para evaluar adecuadamente esta progresión.
La imagen en EH de inicio en la edad adulta ha sido bien descrita, lo contrario ocurre en pacientes con EHJ. Ante esta limitante se han publicado diversos estudios tratando de ofrecer un apoyo al clínico. Vicent y cols (29) reportaron en seis pacientes de EHJ, todos presentaron atrofia de núcleos caudados. Asimismo, Gonzalez-Alegre y Afifi (22) evidenciaron en 12 pacientes con EHJ la presencia de atrofia de caudados y putamen a los dos años de inicio de síntomas. Schapiro y cols (30) también mostraron atrofia de caudados en dos casos de variante Westphal. La mayoría de los pacientes en la serie presentada presentaron atrofia. Esta anormalidad es similar no solo a lo ya reportado en casos juveniles, sino también a los hallazgos característicos de casos adultos con EH. En resumen, los casos EHJ presentan los mismos hallazgos en resonancia magnética estructural que otras variantes de EH por lo que su utilidad clínica es limitada.
La mayoría de nuestros pacientes recibían tratamiento para la sintomatología motora, seguido de antidepresivos. En un estudio realizado en Reino Unido (31) se encontró diferencia en cuanto a la frecuencia de prescripción de estos pacientes en los cuales en dicho estudio mostraron mayor indicación para tratamiento antidepresivo que manejo para alteraciones motoras (66.6%, sobre 58.3%).
En la Tabla 2 se presentan las características principales de las publicaciones a nivel mundial referente a la frecuencia de EHJ en muestras de pacientes con EH. Solo un estudio reportó una prevalencia superior a lo esperado, esto puede deberse que en dicho estudio estuvo involucrado un servicio de neuropediatría (26).
Tabla 2 Prevalencia de casos de enfermedad de Huntington juvenil reportada en la literatura mundial.
| Fuente | Período de inclusión | N. pacientes total | N. pacientes inicio juvenil/infantil | País | Prevalencia EHJ 1 (%) |
|---|---|---|---|---|---|
| Morrison et al. (32) | 1920-1991 | 101 | 10 | Irlanda del norte | 10 |
| Almqvist et al. (33) | 1993-2000 | 205 | 2 | ND2 | 0.9 |
| Siesling S, et al. (34) | ND2 | 2,787 | 65 | Países bajos | 2.3 |
| Alonso M, et al. (6) | 1973-2008 | 691 | 57 | México | 8.2 |
| Mabel E, et al. (26) | 2003-2013 | 71 | 14 | Argentina | 19.7 |
| Panas M, et al. (18) | 1995-2008 | 248 | 13 | Grecia | 5.24 |
| Hayden M, et al. (17) | ND2 | 480 | 17 | Sur de África | 7.7 |
| Gonzalez-Alegre, et al. (22) | 1988-2002 | ND | 12 | Estados Unidos | ND |
| (2019) | 2011-2018 | 198 | 13 | México | 6.5 |
1 enfermedad de Huntington juvenil (EHJ). 2 no disponible (ND)
El presente estudio tiene limitaciones importantes. La población seleccionada fue valorada y tratada en un centro de referencia de tercer nivel por lo que existe un sesgo inherente. Los datos fueron obtenidos del expediente clínico.
CONCLUSIONES
La prevalencia de casos juveniles en una población mexicana fue de 6.5% y el fenotipo motor de estos pacientes fue corea en poco más de la mitad de los casos.
Todos los pacientes de la muestra presentaron deterioro cognitivo desde leve hasta demencia subcortical. Se requieren estudios de cohorte para poder valorar la progresión.
El 84.6% presentó anormalidades en RM y todas ellas corresponden a características bien identificadas en el inicio adulto.
Nuestro trabajo provee un aporte adicional a lo publicado en México, e intenta sumarse a los trabajos ya realizados en otros centros a nivel mundial, con el fin de representar una perspectiva epidemiológica y descriptiva de una población particular de afectados.