









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las Mucopolisacaridosis (MPS) son un grupo de enfermedades que resultan de la deficiencia de enzimas lisosomales que normalmente degradan los glucosaminoglicanos (GAG) caracterizadas por la acumulación lisosomal de sustancias intermedias del metabolismo de los mucopolisacáridos o GAG (Feillet, wiedemann, Jeannesson, Jaussaud & Journeau, 2016) los cuales tienen entre una de sus funciones dar soporte estructural a los tejidos. El tejido conectivo es el que produce más GAG, aunque en menor cantidad se forman en otras células como hepatocitos, células renales, córnea, mastocitos y en la pared vascular. Se identifican cuatro tipos diferen- tes de glucosaminoglicanos, todos sulfatados: heparán, dermatán, queratán y condroitín, que son degradados por 11 diferentes enzimas (ruiz, Hernández, & rosa, 2014).
La primera descripción de un caso de MPS, entre los años 1900 y 1913, se atribuye a John Thompson, de Edimburgo, mientras que la primera publicación sobre este grupo de enfermedades fue realizada en 1917 por Charles Hunter (Mabe, 2004).
Hasta el momento se han descrito siete tipos de MPS: MPS I o síndrome de Hurler (variantes Hurler, Hurler-Scheie y Scheie), MPS II o síndrome de Hunter (variantes A y B), MPS III o síndrome de Sanfilipo (variantes A-D), MPS IV o síndrome de Morquio (variantes A y B), MPS VI o Marotaux-Lamy, MPS VII o síndrome de Sly, MPS IX o síndrome de Natowicz y las deficiencias de múltiples sulfatasas (Suarez-Guerrero, Gómez Higuera, Arias Flórez & Contreras-García, 2016). Determinados por diferentes defectos enzimáticos, pero con características fenotípicas semejantes entre ellos. La incidencia global de las MPS se es- tima en 1:10.000 a 1:25.000 recién nacidos vivos. Sin embargo, esta cifra probablemente es una subestimación de la incidencia real. La MPS III se considera el tipo más frecuente, siendo la tipo VII y la descrita recientemente tipo IX las que se han diagnosticado con menor frecuencia (Mabe, 2004). Aunque la MPS tipo I parece ser una de las más frecuentes en Brasil y la más frecuente en los países de Escandinavia (Colombo, Cornejo & raimann, 2017).
Todas las mucopolisacaridosis tienen herencia autosómica recesiva, excepto el Síndrome de Hunter (MPS II) cuya herencia es recesiva ligada al X. Sin embargo, existen pacientes de sexo femenino reportadas con mucopolisacaridosis II (Colombo et al, 2017).
Se considera a la MPS I como el prototipo de este tipo de enfermedades y junto con la MPS II, así como la VI son las que hasta el momento reciben terapia de reemplazo enzi- mático (ruiz, Hernández & rosa, 2014).
Las diferencias entre los diversos fenotipos dependen parcialmente del tipo de sustrato acumulado, pero también del grado de deficiencia enzimática, del genotipo y de otros factores hasta ahora desconocidos, que podrían explicar cuenta de la baja correlación fenotipo-genotipo observada en algunas series de la literatura. Síntomas comunes a todas las MPS son el compromiso multisistémico con curso crónico y progresivo, que afecta especialmente al sistema esquelético y cardio-pulmonar, a la piel y los faneros, la córnea, el hígado y el bazo (Mabe, 2004).
Presentan una amplia gama de manifestaciones clínicas, entre las que destacan compromiso neurológico, facies tosca, visceromegalias, alteraciones óseas y afección al sistema auditivo, visual, cardiovascular y locomotor (ruiz et al., 2014).
Debido a la gran variabilidad de las manifestaciones clínicas en los pacientes afectados, el diagnóstico usualmente es difícil y puede pasarse por alto, lo que suele conducir a la subestimación de la frecuencia de algunos ti- pos más sutiles clínicamente. Otro problema asociado es el hecho de que no existe un centro de referencia para el diagnóstico y manejo de estas enfermedades, por lo que hay un gran número de instituciones diferentes responsables de estos pacientes y se hace extremadamente difícil acceder a todos los registros (Gómez, García-robles & Suárez-Obando, 2012).
Los fenotipos clínicos no se pueden distinguir bioquímicamente por técnicas de diagnóstico de rutina, ya que todos presentan el mismo comportamiento; el análisis mutacional permite clasificar a algunos pacientes; pero, básicamente, la clasificación se realiza por criterios clínicos (Menéndez-Sainz, Zaldívar-Muñoz & González Quevedo, 2003).
En Ecuador no existen cifras oficiales del porcentaje nacional de los pacientes con enfermedades raras, es un tema poco desarrollado aún, aunque ya se miran avances en el mismo, dado que ya se encuentra dentro de la Ley Orgánica reformatoria a la Ley Orgánica de Salud, Ley 67, que indica la prioridad que los pacientes de enfermedades raras tienen para la disposición médica, lo que aún no garantiza que todos los pacientes sean atendidos, entre éstas están las enfermedades lisosomales y ese problema dificulta su estudio y tratamiento (rueda-Villacis, 2015).
En consecuencia, con esta investigación se pretende contribuir con la caracterización clínica de pacientes con MPS en la provincia de Manabí, así como la identificación de los tipos de MPS existentes en dicha provincia. Datos que serán de gran utilidad para gran parte de la comunidad médica, favoreciendo en un futuro la mejor comprensión de este tipo de enfermedades lisosomales, así como su diagnóstico y tratamiento.
METODOLOGÍA
Se realizó un estudio cualitativo, exploratorio y transversal en el área de consulta externa de la especialidad de Genética del Hospital ¨Dr. Verdi Cevallos Balda” de la ciudad de Portoviejo, provincia Manabí, zonal 4, Ecuador, octubre 2017 - enero 2018.
La población estuvo constituida por los pacientes que constaban en la base de da- tos de la especialidad de Genética Clínica del Hospital ¨Dr. Verdi Cevallos Balda¨. La muestra fue no probabilística por conveniencia, conformada por 30 pacientes que constaban en el registro de la consulta de genética.
Criterios de inclusión.
Pacientes con diagnóstico clínico de mucopolisacaridosis que pertenezcan a la zonal 4 de salud.
Criterios de exclusión.
Pacientes con diagnóstico clínico de mucopolisacaridosis que no pertenezcan a la zonal 4 de salud.
Recolección de la información.
La recolección de datos de los pacientes que acudieron a la consulta de genética del Hospital ¨Dr. Verdi Cevallos Balda¨, procedentes de la ficha médica de cada paciente con diagnóstico de mucopolisacaridosis.
Los datos que se registraron fueron: tipo clínico de MPS, y las características clínicas por regiones y sistemas.
Los datos recolectados fueron tabulados empleando medidas de frecuencia (porcentajes), y su presentación en gráficos.
RESULTADOS Y DISCUSIÓN
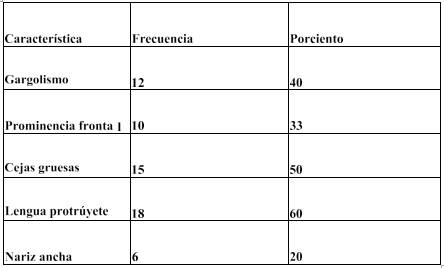
En la Tabla 1 (Ver Anexos) se observa que de acuerdo a las características clínicas que presentan los pacientes con mucopolisacaridosis, en la región de la cara, el rasgo dismórficos que demostró el mayor porcentaje fue la lengua protruyente con un 60%, seguido de cejas gruesas con 50%. Mientras que los rasgos dismórficos que se presentaron en menor porcentaje fueron gargolismo 40%, prominencia frontal 33%, nariz ancha 20%.
La literatura refiere que entre las manifestaciones clínicas frecuentes en las MPS se incluyen la facie tosca, el cabello y cejas gruesos datos que se corresponden con lo descrito en esta investigación. Sin embargo, podemos acotar que en este estudio se encontraron rasgos dismórficos no descritos en la literatura, por lo cual esta investigación estaría aportando nuevos rasgos no mencionados hasta el momento, como lo son la lengua protruyente y gargolismo.
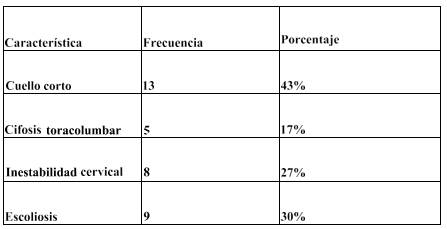
Tabla 2. Características clínicas según alteraciones en columna vertebral. Fuente:Ficha Médica. (Ver Anexos)
En Tabla 2 se observa que de acuerdo a las características clínicas que presentan los pacientes con mucopolisacaridosis, en la región de la columna, el cuello corto, demostró obtener el mayor porcentaje de afectación con un 43%, seguido de escoliosis con 30%. Mientras que en un porcentaje menor obtuvimos 27% con inestabilidad cervical, cifosis toraco- lumbar 17%, y ningún paciente con vertebras aplanadas.
Los síntomas comunes a todas las MPS son el compromiso multisistémico con curso crónico y progresivo, que afecta especialmente al sistema esquelético (Mabe, 2004) dato que tiene relación con la publicación realizada por la Universidad Nacional de Colombia Facultad de Medicina, donde en un estudio clínico en 27 individuos con síndrome de Morquio A
, un 70% de ellos presentó escoliosis (Tapiero, 2016).
También se reportó un caso de un paciente masculino de 19 años de edad, con diagnóstico confirmado de MPS I (variedad Hurler-Scheie) y tratamiento enzimático con respuesta clínica favorable, éste presentó cuello corto con imposibilidad para la flexión-ex- tensión, pectus excavatum, escoliosis y placas a nivel lumbar y cervical (Pineda-Galindo & Moranchel-García, 2015).
En la Tabla 3 (Ver Anexos) se observa que de acuerdo a las características clínicas que presentan los pacientes con mucopolisacaridosis, en la región del tórax, clavículas pequeñas y engrosadas demostró obtener el mayor porcentaje de afectación con un 40%, seguido de escapulas engrosadas y elevadas con un 30%. El menor porcentaje lo ocupa costillas horizontalizadas con 23%.
Como afirma Tapiero (2016) los síntomas iniciales más frecuentes son la deformidad torácica en un 48,15%.
Dados los resultados de la presente investigación, se puede afirmar que, independientmente de cuál sea el tipo de mucopolisacaridosis que se encuentre afectando al individuo, este siempre evolucionará presentando cualquier deformidad torácica en el trayecto de su evolución.
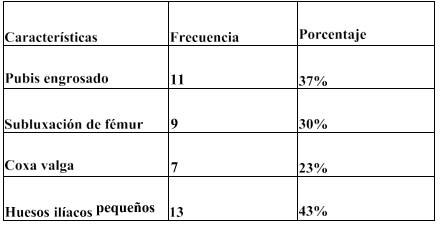
En la Tabla 4 (Ver Anexos) se observa que de acuerdo a las características clínicas que presentan los pacientes con mucopolisacaridosis en la región de la pelvis y cadera, se demostró que el mayor porciento de afectación se constató en huesos ilíacos pequeños, con un 43%, seguido de pubis engrosado con un 37%. Los porcentajes menores lo ocuparon subluxación de fémur 30% y coxa valga 23%.
Dato que se relaciona con un estudio realizado en Valencia, España, donde se revisaron una serie de 28 pacientes con diagnóstico establecido de MPS, encontrando: 8 tipo I (enfermedad de Hurler), 11 tipo II (enfermedad de Hunter), 2 tipo III (enfermedad de San Filippo), 7 tipo IV (enfermedad de Morquio), Los hallazgos radiológicos comunes detecta- dos en todas estas MPS a nivel de pelvis, fue el estrechamiento de la parte inferior de los huesos ilíacos con alas anchas, displasia de la epífisis femoral proximal y coxa valga. Este estudio concluyó afirmando que conocer los hallazgos radiológicos principales de las MPS permite enfocar el diagnóstico para orientar al estudio metabólico y genético, con el fin de iniciar de manera temprana el tratamiento ( Guasp Vizcaíno, Gómez, Mainegra & Monte- sinos,2014).
En cuanto a nuestra experiencia investigativa podemos afirmar que, en la citada región anatómica, los pacientes con mucopolisacaridosis presentan uno de los mayores inconvenientes, el cual es además incapacitante. Los niños que presentaban una evolución corta de la enfermedad, podían caminar con casi ninguna o poca dificultad, mientras que los que presentaron una evolución más avanzada tenían que usar sillas de rueda o ser cargados por sus cuidadores, lo cual nos llevó a deducir que este problema no sucedería si hubiera una detección temprana y a la vez un tratamiento oportuno que estuviera al acceso de quienes padecen esta enfermedad.
En el Gráfico 1 (Ver Anexos) se observa que de acuerdo a las características clínicas que presentan los pacientes con mucopolisacaridosis, en el sistema neurológico, el retraso del lenguaje demostró el mayor porcentaje de afectación con un 73%, seguido del déficit de atención y discapacidad intelectual, ambos con un 63%, el retraso motor en un 60% y en un porcentaje menor se presentó la regresión del desarrollo 53%, e insomnio con 47%.
Sin embargo, en estudio realizado por Tapiero (2016) el 92,6% de los pacientes con MPS presentaron trastornos de la marcha, correspondiéndose con el retraso motor que se evidenció en nuestra investigación.
Podemos señalar que el retraso del desarrollo juega un papel incapacitante desde su temprana aparición y que a una corta edad es difícil su detección, en este caso es imprescindible una historia clínica bien realizada y exhaustiva, y solo así puede ser detectado el problema a tiempo. En los casos objeto de nuestra investigación, la mayoría de las madres refirieron que sus hijos presentaban dificultad al lactar, a la hora de realizar la succión, lo cual fue motivo para acudir a la consulta.
En la literatura se define que la MPS III, Síndrome de Sanfilippo, está marcado por síntomas neurológicos graves, tales como demencia progresiva, comportamiento agresivo, hiperactividad, convulsiones, algo de sordera y pérdida de la visión y una incapacidad para dormir por varias horas seguidas. Este trastorno tiende a presentar tres etapas principales. Durante la primera etapa, el desarrollo inicial de las capacidades mentales y motoras puede presentar algún retraso. Los niños afectados muestran un deterioro significativo en el aprendizaje entre las edades de 2 y 6 años, seguida por la pérdida eventual de capacidades de lenguaje y la pérdida parcial o total de la audición. Algunos niños nunca aprenden a hablar. En la segunda etapa del síndrome, el comportamiento agresivo, la hiperactividad, la demencia profunda y el sueño irregular hacen que estos niños sean difíciles de manejar, particularmente los que poseen una fuerza física normal. En la última etapa del síndrome, el mantenerse en pie se hace cada vez más difícil para estos niños y la mayoría deja de caminar a la edad de 10 años (National Institute of Neurological Disorders and Stroke, 2016).
El síndrome provoca síntomas neurológicos considerables, como discapacidad intelectual severa, y coeficiente de inteligencia puede estar por debajo de 50. La mayoría de las personas con este síndrome viven hasta los años de adolescencia, algunos pueden vivir más, mientras que otros con formas severas de la enfermedad mueren a una edad más temprana. Los síntomas parecen más severos en personas con el síndrome de Sanfilippo tipo A (Servin, Manuel, Cesy & Lafuente, 2014).
En el curso de la Mucopolisacaridosis tipo III, los pacientes pueden presentar además hipoacusia de transmisión, no es inhabitual que el compromiso del lenguaje se atribuye exclusivamente a este hecho, lo que retrasa el diagnóstico de MPS III o enfermedad de Sanfilippo (Mabe, 2004).
En nuestro estudio pudimos concluir que el déficit de atención, el cual fue el segundo mayor por ciento, siempre tiene una relación con el trastorno del sueño, podemos presumir que justamente el 47% de nuestra muestra puede estar atravesando la segunda etapa indicada.
En el Gráfico 2 (Ver Anexos) se evidencia que entre los distintos tipos de mucopolisacaridosis, el tipo III se encuentra presente en el 53% de los pacientes, seguido del tipo II con un 40%, y en menor proporción la mucopolisacaridosis tipo VI con un 7%.
Estos datos coinciden con lo reportado en la literatura, donde se afirma que la MPS III se considera el tipo más frecuente con una prevalencia de 0.28-4.1 por cada 100 000 na- cimientos.(ruiz et al., 2014), siendo la tipo VII y la descrita recientemente tipo IX las que se han diagnosticado con menor frecuencia (Mabe, 2004).
La MPS tipo III o síndrome de Sanfilippo, se divide en 4 tipos, dependiendo del tipo de déficit enzimático que presenta, afectando el metabolismo del heparán sulfato, también se trata de una enfermedad autosómica recesiva, en la que sus síntomas se centran fundamentalmente a nivel de sistema nervioso central. Se manifiesta con características faciales específicas, además de retraso del desarrollo psicomotor, alteraciones cognitivas, ciclo de sueño vigilia, agresividad, hiperactividad y además conducta autista (Jury Hernández, 2013).
Sin embargo, cabe señalar que un estudio de estimación de las frecuencias de las mucopolisacaridosis y análisis de agrupamiento espacial en los departamentos de Cundinamarca y Boyacá (Colombia), la frecuencia combinada para todas las mucopolisacaridosis fue de 1,98 casos por 100.000 nacidos vivos. La mayor frecuencia fue para la de tipo IV, con 0,68 casos por 100.000 nacidos vivos, mientras que la III fue la menor, con 0,17 casos (Gómez et al., 2012).
La MPS tipo II o Síndrome de Hunter, se debe al déficit enzimático de la irunidato-2-sulfatasa. A diferencia de las otras MPS, este es un trastorno que afecta principalmente a hombres jóvenes, ya que tiene una herencia ligada al cromosoma X y sólo hace algunos años se han descrito formas que afectan a mujeres. Su incidencia es 0,31 a 0,71 por cada
rN vivos (Hernández, 2013).
Hasta hace poco la enfermedad de Hunter se había descrito como moderada o grave. Sin embargo, hoy en día, a partir de lo que se conoce sobre la enzima y su gen, está claro que abarca un amplio espectro de gravedad. Algunos afectados experimentarán un retraso progresivo de su desarrollo y un aumento progresivo de problemas físicos graves, otros tendrán una inteligencia normal y problemas físicos progresivos, en algunos los efectos serán más graves que en otros. Es importante recordar que la enfermedad de Hunter tiene efectos sumamente variados. La incidencia de la MPS II se calcula según el registro euro- peo de las Mucopolisacaridosis y Síndromes relacionados, que afecta a 1 de cada 100.000 varones recién nacidos (Ligado al cromosoma X), aunque hay reportes muy escasos en niñas ( Pineda, Vilaseca y Pérez López, 2012).
Los estudios epidemiológicos en la mucopolisacaridosis tipo VI son limitados. La frecuencia relativa en comparación con otras mucopolisacaridosis se ha reportado en don- de ha sido posible, los rangos van del 2-4% de todas las mucopolisacaridosis en Escandinavia hasta el 18.5% en Brasil. (Valayannopou- los, Nicely, Harmatz, & Turbeville, 2010). Por ser una enfermedad de baja prevalencia es frecuente que la comunidad médica no esté familiarizada con las manifestaciones clínicas de la mucopolisacaridosis tipo VI, por lo que el diagnóstico se vuelve un reto para el clínico. El retraso en el diagnóstico de éstos pacientes origina un deterioro multiorgánico progresivo, que puede llevar al paciente a la postración y falla severa de órganos incluso en la primera década de la vida. Para su manejo, se requiere de un equipo de salud multidisciplinario, con conocimiento y experiencia en la patología (Arellano y Ávila rejón, 2010).
CONCLUSIONES
Los rasgos dismórficos más frecuentes en los pacientes con MPS, a nivel de la cara, fueron lengua protruyente y cejas gruesas.
En relación a los aparatos y sistemas se evidenció una alta incidencia de pacientes con cuello corto, clavículas pequeñas y engrosadas, huesos iliacos pequeños y retraso del lenguaje.
De acuerdo a las características clínicas de la población de estudio, el tipo más frecuentes de mucopolisacaridosis fue el tipo III, seguida por el tipo II, y en último lugar el tipo VI.