









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
1. INTRODUCCIÓN
El coronavirus denominado SARS-CoV-2 causante de la enfermedad COVID-19 fue reportado por primera vez en diciembre de 2019 en la ciudad de Wuhan, en la P.R. China (Xu y col., 2020) y fue declarado pocos meses después pandemia por la Organización Mundial de la Salud (OMS). Este virus para principios de mayo de 2020 ya había causado más de 4.000.000 de casos confirmados y más de 250.000 muertes a nivel mundial (WHO, 2020). Lo que ameritó una acelerada carrera para la búsqueda de una vacuna no disponible aún para la fecha (Jiang y col., 2020). Sin embargo, diversos especialistas han recomendado considerar drogas previamente aprobadas por la FDA para ser estudiadas como potenciales agentes alternativos de tratamiento, incluyendo diversos compuestos naturales ampliamente conocidos (Chang y col., 2020; Contini, 2020). De hecho, se han realizado amplios estudios teóricos y bioinformáticos para predecir la potencialidad de diversos compuestos fitoquímicos dirigidos al acoplamiento molecular y a la perturbación estructural de proteínas estructurales y funcionales del SARS-CoV-2 (Khaerunnisa y col., 2020; Qamar y col., 2020; Sharma y Kaur, 2020; Chandel y col., 2020; Adem y col., 2020; Gentile y col., 2020; Sun y col., 2020a,b) siendo la RNA polimerasa una de las dianas de mayor interés terapéutico (Chang y col., 2020; Elfiky, 2020a; Gordon y col., 2020).
Sin embargo, los estudios computacionales destinados a predecir los mejores acoplamientos moleculares se han centrado en el uso del algoritmo y función de puntuación AutoDock Vina (Chang y col., 2020; Zhang y col., 2020; Chandel y col., 2020; Sekhar, 2020; Gentile y col., 2020; Alamri y col., 2020). AutoDock Vina es una nueva función de puntuación basada en el conocimiento que utiliza una variante de X-Score, con un ajuste usando PDBbind, y considerando además las contribuciones inter e intramoleculares, su técnica de muestreo se basa en el optimizador global de Búsqueda Local Iterativa, en su variante Broyden - Fletcher - Goldfarb - Shanno (BFGS) (Pagadala y col., 2017). En este sentido, además del uso del AutoDock Vina, en el siguiente estudio nosotros proponemos realizar un análisis comparativo entre los acoplamientos de diversos algoritmos incluidos en el paquete Molegro Molecular (Thomsen y Christensen, 2006). Específicamente, consideramos a MolDock el cual es un nuevo algoritmo híbrido de búsqueda heurística que combina la evolución diferencial con un algoritmo de predicción de cavidades, y aplica como función de puntuación una extensión del Potencial Lineal por Partes (PLP) que considera la direccionalidad, cargas del puente de hidrógeno e interacciones electrostáticas, y ha demostrado ser una de las más precisas para cribados rápidos porque las poses mejor clasificadas son re-clasificadas mediante un re-ranking que utiliza una función de puntuación más compleja que agrega un término de torsión sp2-sp2 al cálculo de puentes de hidrógeno y un término potencial de Lennard-Jones a la puntuación (Torktaz y col., 2013; Naeem y col., 2013).
En paralelo se calculó el algoritmo Rerank también integrado en el Molegro, que se basa en funciones de clasificación de tipo Relación cuantitativa Estructura-Actividad (QSAR, por sus siglas en ingles), el cual representa una función muy costosa para su cálculo en medio o durante la simulación de acoplamiento, pero que es generalmente mejor que la función de puntuación de acoplamiento por defecto para rectificar o determinar la mejor pose entre ciertas poses que emergen del ligando. En Molegro la función Rerank proporciona una evaluación del poder de las interacciones, y tiende a ser exitoso al reorganizar varias posturas de un mismo ligando (Velraj y Chand, 2019).
Por otro lado, para adquirir u obtener una evaluación aproximada de las poses mejor clasificadas, se usó una estimación de afinidad vinculante, por lo que también fue considerado el algoritmo PLANTS (Protein-Ligand ANT System), que se fundamenta en un algoritmo de optimización estocástica llamado optimización de colonias de hormigas (ACO, por sus siglas en inglés) y cuya función de puntuación empírica está basada de igual manera en un PLP similar al utilizado por MolDock, pero integra más tipos de interacción (repulsivas, atractivas, no polares, enlaces de hidrógeno y metálico) por lo que al ser más conservador que MolDock, se ha recomendado como estrategia de re-ranking posterior (Torktaz y col., 2013).
También comparamos los acoplamientos arrojados por el algoritmo DockT del servidor DockThor, el cual implementa un método basado en la cuadrícula que emplea un algoritmo genético de estado estable para múltiples soluciones como motor de búsqueda y el campo de fuerza MMFF94S como la función de puntuación para la evaluación de pose. Esta función de puntuación se obtiene mediante regresión lineal y técnicas de aprendizaje automático más sofisticadas para problemas no lineales utilizando el conjunto refinado PDBbind (Da Silveira y col., 2019; Dos Santos y col., 2020).
Todas estas funciones empíricas anteriormente descritas coinciden en estar apoyadas en análisis de regresión que usan diferentes términos para describir contactos vdW, cobertura superficial lipofílica, enlaces de hidrógeno, desolvatación, tensión y flexibilidad de ligando (Pecina y col., 2016) para evaluar y clasificar con mayor precisión las poses predichas utilizando una función de puntuación. Sin embargo, como fue definido, las cinco estrategias de acoplamiento consideradas en este estudio, poseen funciones de puntuación con algunas divergencias. En este sentido, proponemos evaluar un grupo destacado de compuestos fitoquímicos con actividad antiviral reportada y dirigidos al bloqueo de la RNA polimerasa del SARS-CoV-2, mediante un análisis computacional comparativo y multivariado de las funciones de puntuación MolDock, Rerank, PLANTS, DockT y AutoDock Vina considerando los términos de solubilidad y liposolubilidad de los ligandos, así como la relación total de hidrofobicidad e índices de hidropatía de las cavidades involucradas en los acoplamientos con la finalidad de aumentar la precisión en la rápida predicción de los mejores acoplamientos durante los estudios teóricos de potenciales compuestos con interés biomédico frente al COVID-19.
2. METODOLOGÍA
Cribado de Acoplamiento Molecular: En este estudio, se consideró la estructura funcional asociada con el SARS-CoV-2 nsp12 Polimerasa (PDB: 6NUR). La cual se obtuvo de la base de datos de proteínas RCSB Protein Data Bank. Y con base en lo descrito en literatura, probamos los siguientes compuestos fitoquímicos con posible actividad antiviral reportada incluidos aquellos con algún tipo de actividad in silico frente el SARS-CoV-2: Capsaicina, Curcumina y Piperina (Chandani y col., 2019; Tang y col., 2020; Bonfim y col., 2020; Gonzalez-Paz y col., 2020), y Epigallocatechin Gallate (EPGG) (Mohammad y col., 2020). Además, hemos utilizado como control de acoplamiento molecular para la RNA polimerasa el medicamento aprobado por la FDA Remdesivir, un “análogo de nucleótido en investigación” (Hendaus, 2020) activo frente a los virus del Ébola, Marburgo, virus sincitial respiratorio, Junín, fiebre de Lassa, MERS, Nipah, Hendra y el SARS, por lo que se ha recomendado para el tratamiento de pacientes con COVID-19 (Al-Tawfiq y col., 2020).
Para esto todas las estructuras 2D de los compuestos se obtuvieron de PubChem (https://pubchem.ncbi.nlm.nih.gov/) en formato SDF, y se utilizó el convertidor en línea SMILES (https://cactus.nci.nih.gov/translate/) para obtener formatos PDB. Para simular la unión ligando-proteína, se construyeron los complejos en formato PDB en el servidor DockThor (https://dockthor.lncc.br/v2/) utilizando el algoritmo de flexibilidad y el acoplamiento ciego que implementa un método basado en la cuadrícula con un algoritmo genético de estado estable para múltiples soluciones, calculando en paralelo la función de puntuación DockT (Da Silveira y col., 2019; Dos Santos y col., 2020). La posición más favorecida se analizó con las herramientas del Molegro Molecular MMV_2019_7.0.0, calculando también las funciones MolDock Score, Rerank Score y PLANTS Score (Thomsen y Christensen, 2006). También se calculó la función de puntuación AutoDock usando el algoritmo AutoDock Vina del servidor DINC 2.0 (Antunes y col., 2017).
Análisis complementarios: el servidor Molinspiration (Cheminformatics, 2020; Reena Roy y col., 2020) y las herramientas del Grupo de Modelado Molecular de la SIB | Instituto Suizo de Bioinformática (Zoete y col., 2016; Daina y col., 2017) se utilizaron para los cálculos de bioactividad. Para el cálculo de hidropatía (GRAVY, por las siglas en inglés de Grand Average Hydropathy Value) y hidrofobicidad total se utilizó el servidor APD3 (http://aps.unmc.edu/AP/prediction/prediction_main.php). Estos parámetros se corroboraron con la herramienta CellPPD, con el sitio WEB dado por https://webs.iiitd.edu.in/raghava/cellppd/index.html. También se aplicó como modelo estadístico el análisis de componentes principales (ACP) para realizar una reducción de variables y establecer previa determinación de co-linealidades (partiendo de correlaciones simples y múltiples según sea el caso) aquellas variables ortogonales que permitan obtener el mayor porcentaje de variabilidad explicada; así mismo se utilizó un modelo multivariado de tipo discriminante (ADC) con las variables resultantes con la finalidad de proponer la función de puntuación de acoplamiento con mayor peso clasificatorio con base a las condiciones de este estudio basado en solubilidad de ligandos y características hidrofóbicas de las cavidades. Todos los análisis se realizaron con el paquete estadístico IBM SPSS-23.
3. RESULTADOS Y DISCUSIÓN
En la Tabla 1, se muestran los resultados del acoplamiento de la Piperina_ID_638024, EPGG_ID_65064, Curcumina_ID_969516, Capsaicina_ID_1548943 y el control Remdesivir_ID_121304016 (ver Figura 1) con la estructura proteica de SARS-CoV-2 nsp12 polimerasa (PDB ID: 6NUR). Estas puntuaciones representan la afinidad de unión en energía libre (ΔG) obtenida con el paquete Molegro Molecular (funciones de puntación MolDock, Rerank y PLANTS) y los servidores DockThor (función de puntuación DockT) y DINC 2.0 (función AutoDock Vina). Todos los algoritmos y sus funciones de puntuación exceptuando AutoDock Vina predijeron que el acoplamiento termodinámicamente más favorable ocurre entre la Piperina y la polimerasa viral. AutoDock Vina fue el único algoritmo que difirió en la predicción del acoplamiento con respecto al resto. Coincidiendo solo en la segunda predicción más probable y favorable termodinámicamente calculada por las otras funciones de puntuación, representado por el acoplamiento entre el fitoquímico EPGG y la polimerasa. Un acoplamiento que ya se ha descrito como favorable (Mohammad y col., 2020) y el cual solo la función de puntuación DockT difirió en la predicción. Adicionalmente, el acoplamiento termodinámicamente más favorecido de la piperina frente al EPGG observado; aquí ya se ha reportado (Tripathi y col., 2020), aunque nuestros resultados difieren de lo predicho por otras funciones de puntuación (Mohammad y col., 2020).
Interesantemente el 80% (4/5) de las funciones de puntuación predijeron que aproximadamente el 50% de los compuestos naturales probados presentan energías de acoplamiento termodinámicamente más favorables que el control Remdesivir. De hecho, ninguna de las funciones de puntuación predijo un acoplamiento de esta molécula más favorable que el resto de los compuestos. Una observación que coincide con el reportado por otros autores para estos compuestos y en relación a otros fármacos controles considerados en sus estudios (Vardhan y Sahoo, 2020). Un hallazgo importante si consideramos que el Remdesivir se ha recomendado para el tratamiento de pacientes con COVID-19 (Al-Tawfiq y col., 2020) y especialmente porque algunos estudios establecen que el Remdesivir no ha aportado beneficios significativos comparado con los placebos (Wang y col., 2020; Grein y col., 2020).
Los acoplamientos más favorables predichos por MolDock, Rerank, PLANTS y DockT fueron de -40.62 kcal/mol, -38.05 kcal/mol, -33.77 kcal/mol y -7.24 kcal/mol para el complejo Piperina-polimerasa, respectivamente; y con AutoDock Vina de -7.40 kcal/mol para EPGG-polimerasa. Lo que representaría una diferencia con respecto al control Remdesivir relativo a cada función de puntuación de aproximadamente 19 kcal/mol, 16 kcal/mol, 4 kcal/mol, 0 kcal/mol para los complejos con Piperina, y de 0.3 kcal/mol para EPGG, respectivamente. Con una diferencia entre el primer y segundo mejor acoplamiento, según sea el caso, de 15 kcal/mol, 9 kcal/mol, 0.8 kcal/mol para la Piperina frente a EPGG, 0.4 kcal/mol para la Piperina frente a Curcumina, y 0.3 kcal/mol para EPGG frente al control. Todos los acoplamientos fueron favorecidos por una media de entre el 85-100% de interacciones estéricas/hidrofóbicas, con residuos que promovían al mismo tiempo interacciones de tipo puentes de hidrógeno. Específicamente, el 100% de las interacciones entre los residuos de la polimerasa y los compuestos EPGG, Capsaicina y Curcumina estuvieron favorecidas por acoplamientos estéricos, con residuos que al mismo tiempo contribuyeron con un 20% para EPGG y Capsaicina, y un 13% para Curcumina de interacciones de tipo puentes de hidrogeno. Mientras que el 94% de las interacciones entre la Piperina y la proteína fueron netamente estéricas con un restante 6% representadas por puentes de hidrógeno. En contraste el fármaco control mostró un 14% de interacciones basadas en puentes de hidrogeno.
El 60% (3/5) de los algoritmos predijeron las interacciones entre EPGG, Curcumina y Capsaicina en la misma cavidad, con una ligera diferencia entre las interacciones calculadas para EPGG y Curcumina, quienes difieren en aproximadamente un par de residuos. Por otro lado, las interacciones predichas para la Piperina, así como para el control Remdesivir, fueron en bolsillos únicos en relación con los residuos involucrados, pero predichas en una misma cavidad ubicada en una región más superficial que la calculada para el resto de los compuestos. Todas las cavidades predichas tienen un tamaño aproximado de ≥ 1700 Å3. Nuestros resultados muestran que, existe una moderada correlación entre la profundidad de la cavidad muestreada (y, por ende, con el tipo y numero de interacciones posibles), y el acoplamiento termodinámicamente más favorable clasificado por todas las funciones de puntuación consideradas en este estudio con un coeficiente Rp = 0.50 (ver Tabla 1).
Es importante señalar que el compuesto GS-441524, es un antiviral análogo de nucleótido de adenosina, similar al Remdesivir (GS-5734), este último tras la administración, se metaboliza en su forma activa GS-441524; por lo tanto, al ser GS-5734 un pro-fármaco del GS-441524 los acoplamientos predichos podrían presentar variaciones, sin embargo, frente a esto, diversos autores justifican el uso del Remdesivir (en lugar del GS-441524) bajo la denominación de “análogo de nucleótido en investigación” por ser una forma con un mejor coeficiente de partición y solubilidad (ver Tabla 2) que la forma activa GS-441524 (log K ow: -0.88 a -1.35; valores predichos con Molinspiration y el Grupo de Modelado Molecular de la SIB). Estas variaciones contribuyen en parte con la falta de consenso a la hora de realizar investigaciones con estos metabolitos (Agostini y col., 2018; Jordania y col., 2018; Siegel y col., 2017; Tchesnokov y col., 2019; Hendaus, 2020). Y a pesar de que probablemente la predicción de los acoplamientos puede verse comprometida por no utilizar la forma activa GS-441524, nosotros encontramos que bajo las condiciones de este estudio la diferencia entre la media termodinámica para el acoplamiento de Remdesivir y GS-441524 con la estructura proteica 6NUR es de aproximadamente -1 kcal/mol (resultados no mostrados) y que se corresponden con lo reportado por otros autores que incluso predicen un acoplamiento más favorable para el Remdesivir (Joshi y col., 2020; Huynh y Luan, 2020). Estas variaciones en la biodisponibilidad, la similitud entre las energías de acoplamiento y otros aspectos que favorecen al Remdesivir (como ser la forma farmacéutica patentada y recomendada por la OMS) son algunas de las razones por las cuales consideramos en este trabajo a Remdesivir (o GS-5734) al igual que lo reportado por otros estudios (Elfiky, 2020a,b; Joshi y col., 2020; Yu y col., 2020). No obstante, recomendamos más estudios teóricos que realicen este tipo de análisis comparativo bajo las condiciones de este estudio.
Tabla 1 Resultados del Cribado virtual con la estructura SARS-CoV nsp12 polimerasa. Se muestran el acoplamiento de los compuestos fitoquímicos considerados y del fármaco control Remdesivir aprobado por la FDA y recomendado para el tratamiento del COVID-19.
| SARS-CoV-2 nsp12 Polimerasa (PDB ID: 6NUR) | ||||||
| Compuesto* | MolDock Score (kcal/mol) | Rerank Score (kcal/mol) | PLANTS Score (kcal/mol) | DockT Score (kcal/mol) | AutoDock Score (kcal/mol) | Interacciones |
| Piperina_ID_638024 | -40.62 | -38.05 | -33.77 | -7.24 | -6.30 | Arg349(A)HB, Leu265(A)SI, Ile266(A)SI, Trp268(A)SI, Ser255(A)SI, Val320(A)SI, Phe321(A)SI, Pro322(A)SI, Pro323(A)SI, Thr319(A)SI, Pro461(A)SI, Asn459(A)SI, Phe396(A)SI, Thr393(A)SI, Cys395(A)SI, Pro677(A)SI |
| EPGG_ID_65064 | -25.95 | -28.95 | -32.96 | -6.81 | -7.40 | Ser814(A)HB-SI, Asp760(A)HB-SI, Ser759(A)HB-SI, Leu758(A)SI, Cys813(A)SI, Asp761(A)SI, Lys621(A)SI, Arg553(A)SI, Asp623(A)SI, Pro620(A)SI, Asp618(A)SI, Tyr619(A)SI, Cys622(A)SI, Thr680(A)SI, Thr687(A)SI, Ser682(A)SI |
| Curcumina_ID_969516 | -20.64 | -25.66 | -22.42 | -6.89 | -6.70 | Asp618(A)HB-SI, Lys798(A)HB-SI, Leu758(A)SI, Asp760(A)SI, Ser814(A)SI, Cys813(A)SI, Asp761(A)SI, Ser759(A)SI, Asp623(A)SI, Cys622(A)SI, Arg553(A)SI, Lys551(A)SI, Tyr619(A)SI, Lys621(A)SI, Pro620(A)SI |
| Capsaicina_ID_1548943 | -25.60 | -22.67 | -26.79 | -6.67 | -6.50 | Asp760(A)HB-SI, Asp618(A)HB-SI, Tyr619(A)SI, Pro620(A)SI, Lys621(A)SI, Arg553(A)SI, Asp623(A)SI, Cys622(A)SI, Ser759(A)SI, Leu758(A)SI |
| Remdesivir_ID_121304016 | -22.17 | -22.47 | -29.85 | -7.24 | -7.10 | Asp454(A)HB-SI, Arg392(A)HB-SI, Ser451(A)SI, Arg457(A)SI, Pro133(B)SI, Asn447(A)SI, Ala30(C)SI, Ser26(C)SI, Leu180(B)SI, Lys27(C)SI, Asp134(B)SI, Gln31(C)SI, Gln34(C)SI, Trp182(B)SI |
H B, puentes de hidrógeno
SI , interacciones estéricas/hidrofóbicas
*Todos los compuestos fueron obtenidos de la base de datos PubChem (https://pubchem.ncbi.nlm.nih.gov/).
Por otro lado, es importante señalar que luego de estudiar las propiedades moleculares y las características estructurales de estos compuestos, se determinó que todos los ligandos fitoquímicos cumplen con todos los criterios establecidos por las reglas de Lipinski garantizando una buena bioacumulación y su posible administración por vía oral en términos de absorción, distribución, metabolismo, y excreción ("ADME") (ver Tabla 2). Exceptuando al EPGG, el cual, sin embargo, a pesar de no cumplir con el número de grupos aceptores y donadores de enlaces de hidrógeno, y al ser características incompatibles con las reglas de Lipinski, esta molécula tiene un buen peso molecular, y un coeficiente de partición < 5, por lo que, aunque es una molécula con algunas dificultades en su permeabilidad, es capaz de bioacumularse con un coeficiente alto de acuerdo con los estándares establecidos (Leo y col., 1971; Ghose y col., 1999; Bhal, 2007). Curiosamente, en términos generales los fisicoquímicos estudiados exhiben mejores características de tipo ADME que las presentadas por el control.
Tabla 2 Cálculo de características consideradas asociadas a la bioactividad y solubilidad de ligandos usando el servidor Molinspiration y las herramientas del Grupo de Modelado Molecular de la SIB | Instituto Suizo de Bioinformática.
| Compuesto | MW (g/mol) | Coeficiente de Partición (log K ow )* | HBA | HBD | Solubilidad (logS) | Liposolubilidad (Log Po/w ) |
|---|---|---|---|---|---|---|
| Piperina_ID_638024 | 285.34 | 3.33 | 3 | 0 | -3.0 / -3.96 | 3.04 |
| EPGG_ID_65064 | 458.38 | 2.25 | 11 | 8 | -2.50 / -4.91 | 1.01 |
| Curcumina_ID_969516 | 368.40 | 3.29 | 6 | 2 | -3.94 / -4.83 | 3.03 |
| Capsaicina_ID_1548943 | 305.40 | 3.04 | 3 | 2 | -3.53 / -4.87 | 3.43 |
| Remdesivir_ID_121304016 | 602.59 | 2.82 | 13 | 4 | -4.12 / -6.01 | 1.50 |
*log K ow , 2 - 5 (Leo et al., 1971; Ghose et al., 1999; Bhal, 2007). Todos los parámetros fueron validados con la base de datos PubChem (https://pubchem.ncbi.nlm.nih.gov/).
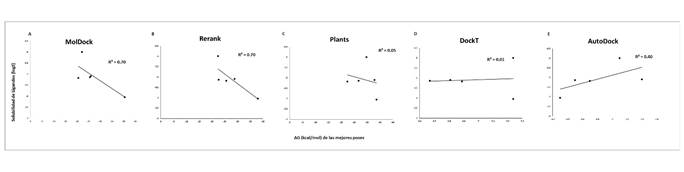
Figura 2. Valores de puntuación de las mejores posiciones calificadas durante los acoplamientos correlacionadas frente al coeficiente de solubilidad de cada ligando. Se muestran las 5 funciones de puntuación consideradas para la evaluación de los 5 ligandos probados frente a la RNA polimerasa del SARS-CoV-2. A: MolDock; B: Rerank; C: PLANTS; D: DockT y E: AutoDock.
Adicionalmente, encontramos que existe una correlación significativa entre la solubilidad del ligando y las funciones de puntuación MolDock y Rerank Rp = 0.70, mientras que para el resto de las funciones de puntuación la correlación fue de débiles a nulas (ver Figura 2). Por otro lado, la función de puntuación AutoDock Vina fue la única que presentó una correlación fuerte con la baja liposolubilidad de los ligandos (Rp = 0.90) (ver Figura 3). En términos de la relación total de hidrofobicidad de las cavidades, encontramos que solo las funciones Rerank y MolDock mostraron una fuerte correlación con esta propiedad siendo esta de Rp = 0.95 y Rp = 0.90, respectivamente. La correlación mostrada por el resto de las funciones fue débil en términos de hidrofobicidad de las cavidades (ver Figura 4). Estos resultados son similares a los arrojados para el término de hidropaticidad con una correlación significativa para MolDock (Rp = 0.82) y perfecta para Rerank (Rp = 0.98) y débil para el resto de las funciones (ver Figura 5). Por lo tanto, se pone en evidencia que las condiciones de acoplamiento propuestas en este estudio aunado al grupo de ligandos considerados, que las calificaciones de los mejores acoplamientos obtenidos a partir de las funciones de puntuación PLANTS, AutoDock Vina y DockT (en menor medida para DockT Score) disminuyen por la solubilidad los ligandos, así como por la relación total de hidrofobicidad e índices de hidropatía de las cavidades. Específicamente, mientras que las funciones de puntuación MolDock y Rerank favorecen a ligandos más solubles acoplados a cavidades más hidrofóbicas, y con una hidropaticidad >0 como en el caso de la piperina. La función de puntuación AutoDock Vina tiende por el contrario a clasificar mejor a los ligandos por menor liposolubilidad acoplados a cavidades con una relación total de hidrofobicidad ≤ 25%, o bien, con una hidropaticidad ≤ -0.89 como la exhibida por las cavidades o bolsillos en los que se acopló el resto de los ligandos considerados en este estudio. Las características consideradas asociadas a la solubilidad e hidropatías de ligandos y proteínas, respectivamente, se detallan en la Tabla 2 y Tabla 3, correspondientes.
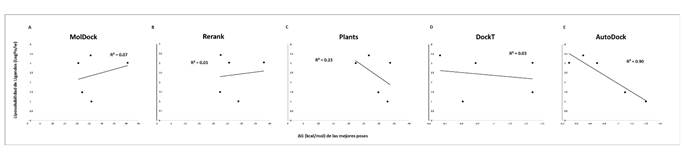
Figura 3 Correlación entre los valores de puntuación de las mejores posiciones calificadas durante los acoplamientos y la liposolubilidad de cada ligando. Se muestran los resultados para las 5 funciones de puntuación consideradas para la evaluación de los 5 ligandos probados frente a la RNA polimerasa del SARS-CoV-2. A: MolDock; B: Rerank; C: PLANTS; D: DockT y E: AutoDock.
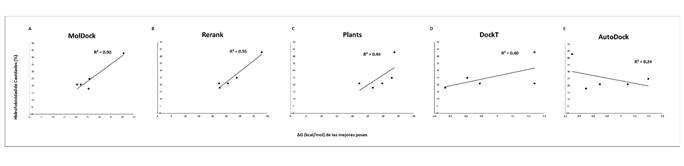
Figura 4. Correlación entre los valores de puntuación de las mejores posiciones calificadas durante los acoplamientos frente a la relación total de hidrofobicidad predicha para las cavidades muestreadas. Se muestran los valores para las 5 funciones de puntuación consideradas para la evaluación de los 5 ligandos probados frente a la RNA polimerasa del SARS-CoV-2. A: MolDock; B: Rerank; C: PLANTS; D: DockT y E: AutoDock.
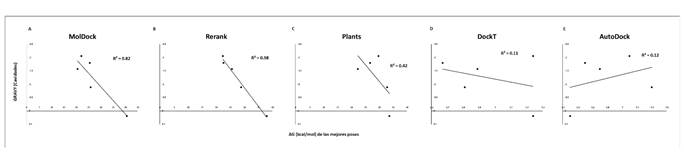
Figura 5. Correlación entre los valores de puntuación de las mejores posiciones calificadas durante los acoplamientos frente al índice de hidropatía (GRAVY) de las cavidades muestreadas. Se muestran los resultados para las 5 funciones de puntuación consideradas para la evaluación de los 5 ligandos probados frente a la RNA polimerasa del SARS-CoV-2. A: MolDock; B: Rerank; C: PLANTS; D: DockT y E: AutoDock.
Tabla 3. Predicción de las características consideradas asociadas a hidropatía (GRAVY) y a la relación total de hidrofobicidad de las cavidades involucradas en los acoplamientos.
| Cavidad o Bolsillo | SARS-CoV-2 nsp12 Polimerasa (PDB ID: 6NUR) | |||
|---|---|---|---|---|
| Localización | Hidrofobicidad (%) | GRAVY | Ligandos Acoplados | |
| 1 | Interna | 18-25 | -0.89 / -1.80 | EPGG / Curcumina / Capsaicina |
| 2 | Externa/superficial | ~20 | ~ -0.06 | Remdesivir |
| 3 | Externa/superficial | ~40 | ~0.19 | Piperina |
GRAVY, siglas en ingles de Grand Average Hydropathy Value.
Este análisis comparativo y la importancia de las divergencias entre el acoplamiento de los ligando se facilitó gracias a que la diferencia entre la relación total de hidrofobicidad del bolsillo predicho para la piperina (fitoquímico mejor clasificado) y del segundo mejor clasificado (EPGG) fue de ≥18%, un margen considerable si se toma en cuenta que la relación total de hidrofobicidad del bolsillo acoplado a uno de los fitoquímicos peor clasificados (Capsaicina) es del aproximadamente 18%. Estas observaciones son coherentes con nuestras simulaciones teóricas por ACP que comprueban la importancia de las características hidrofóbicas de la cavidad, así como las del ligando para establecer las interacciones más favorecidas en la predicción de un complejo, y agrupan a las variables según el aporte en magnitud al componente 1 que explica aproximadamente el 55% del fenómeno de la siguiente manera: Rerank > MolDock > PLANTS > DockT, incluyendo a la función AutoDock Vina en el componente 2 responsable de un aproximadamente 32% del peso total de la variabilidad explicada (ver Figura 6a). Un resultado similar al obtenido por el modelo ADC el cual predijo que la funciones con mayor peso clasificatorio o predictivo para la categorización de la relación entre las características de hidrofobicidad y las funciones de puntuación podrían ordenarse de la siguiente manera: MolDock > Rerank > PLANTS excluyendo a DockT y AutoDock Vina (ver Figura 6b). Ambos modelos coinciden en el peso explicativo, predictor y clasificatorio de las funciones Rerank, MolDock y PLANTS.
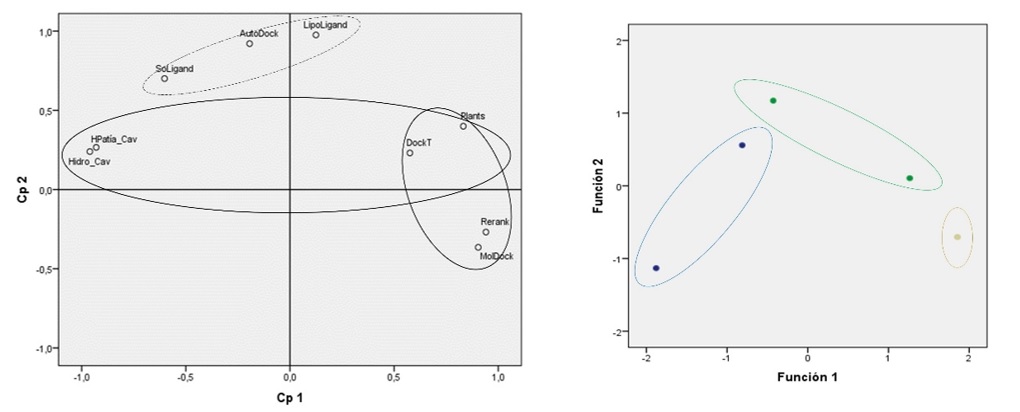
Figura 6 A) Análisis de componentes principales entre las variables consideradas en este estudio incluidas las funciones de puntuación, y las basadas en solubilidad de ligandos y características hidrofóbicas de las cavidades. Se muestra el componente principal numero 1 (Cp1) el cual agrupa a las funciones de puntuación MolDock, Rerank, PLANTS y DockT, y a las variables asociadas a la hidrofobicidad de las cavidades (puntos agrupados en los anillos negro-solido); y al componente 2 (Cp2) que agrupó a la función AutoDock y a las variables relacionadas con la solubilidad del ligando (puntos agrupados en el anillo negro-punteado). Estos componentes 1 y 2 (o variables latentes) generados por la agrupación de las variables primarias permiten explicar el 55% y 32% de la variabilidad observada; B) Análisis discriminante entre las variables consideradas en este estudio que incluye todas las funciones de puntuación, la solubilidad de ligandos y las características hidrofóbicas de las cavidades. La función discriminante con mayor peso clasificatorio o predictivo para la categorización de la relación entre las características de solubilidad e hidrofobicidad y las funciones de puntuación está representada por MolDock (función 1, que agrupa los puntos de color azul) y Rerank (función 2, que agrupa los puntos de color verde). Estas funciones discriminantes permitieron clasificar correctamente el 100% de las observaciones bajo las condiciones de nuestro estudio.
En este sentido, estos resultados preliminares sugieren que la eficacia de las funciones de puntuación, para una correcta clasificación de los acoplamientos obtenidos bajo las condiciones de muestreo de este estudio, y en concreto para aquellos ligandos solubles con átomos o grupos químicos que puedan establecer enlaces tanto polares como no polares acoplados a cavidades hidrofóbicas, se puede ordenar de la siguiente manera: [Rerank > MolDock > PLANTS > AutoDock Vina > DockT]. Esta categorización es producto de las predicciones de los modelos ACP, ADC y del análisis de los coeficientes Rp discutidos en este estudio, y son similares a los reportados por otros autores cuyos análisis de correlación entre poses experimentales y teóricas ordenan las funciones de puntuación de la siguiente manera: PLANTS > MolDock > AutoDock Vina según Pearson, y MolDock > PLANTS > AutoDock Vina según Spearman (Xu y col., 2015). Resultados consistentes con la posición relativa obtenida en nuestro estudio para MolDock en relación a AutoDock Vina.
Estos resultados son relevantes debido a que se ha reportado sobre la importancia del efecto de la hidrofobicidad del bolsillo de unión, y se ha descrito el rendimiento de algoritmos híbridos empíricos como Glide que son mejores para los bolsillos de unión hidrofóbicos o hidrófilos que para los bolsillos con hidrofobicidad intermedia, mientras que otros algoritmos genéticos con funciones de puntuación basadas en campo de fuerza y PLP como Gold, tienden a funcionar mejor solo con los bolsillos hidrofóbicos. Además, se ha señalado que estos algoritmos en términos generales tienen un rendimiento relativamente mejor para ligandos hidrófobos pequeños (Chaput y Mouawad, 2017). Por lo tanto, no hay una función de puntuación o protocolo en particular que pueda recomendarse exclusivamente para proteínas o ligandos específicos, y aunque algunos programas se han sugerido por funcionar globalmente mejor que otros, como Glide y Gold (Chaput y Mouawad, 2017), curiosamente, otros autores han evaluado la precisión en la predicción del acoplamiento de MolDock y han obtenido mejores resultados que los arrojados precisamente por Glide y GOLD (Thomsen y Christensen, 2006).
Consecuentemente, debido a los más de 160 algoritmos de acoplamiento desarrollados y a las más de 109 funciones de puntuación descritas (Rawal y col., 2019), y dada la diversidad bioquímica de ligandos y proteínas, nosotros proponemos con base en la evidencia y a los resultados obtenidos en este estudio, el uso del algoritmo híbrido MolDock para cribados rápidos y al Rerank para reorganizar la clasificación de los acoplamientos obtenidos cuando se trabaje con ligandos solubles como la piperina, indistintamente de la polaridad, y que estén dirigidos a cavidades hidrofóbicas como las de la RNA polimerasa viral del SARS-CoV-2, especialmente para los enfoques computacionales en el contexto del desarrollo rápido de fármacos. También recomendamos validar la reproducibilidad de estos resultados con un número mayor de ligandos y proteínas, con la finalidad de proponer el desarrollo y aplicación de un algoritmo de reclasificación basado en características hidrofóbicas de los complejos ligando-proteína y que funcione de manera genérica acoplado a las funciones de puntación ya descritas derivadas de los algoritmos de acoplamiento, para continuar con la optimización de la discriminación de los mejores fármacos durante la investigación y desarrollo de candidatos en las etapas in silico.
Adicionalmente, para validar el impacto que tengan otros aspectos no considerados como las perturbaciones estructurales que podrían ser inducidas en la proteína por los ligandos estudiados sobre las recalificaciones obtenidas con las funciones de puntuación propuestas, nuestra investigación contempla una segunda etapa incorporando variables asociadas a la dinámica molecular (DM) de los complejos derivados, proponiendo realizar simulaciones de acoplamiento con tres propósitos: (1) estudiar la estabilidad relativa del ligando que reside en la cavidad de unión; (2) muestrear las conformaciones de mínima energía para calcular la perturbación de la estabilidad termodinámica y estructural de los complejos; y (3) analizar la distribución de cavidades y los coeficientes de deformación estructural en las conformaciones de mínima energía muestreadas. Para esto es importante modelar sistemas basados en una copia de la polimerasa viral, una copia del ligando acoplado y aproximadamente 50 iones Na+ y Cl- para simular condiciones fisiológicas. Todo el sistema tiene que ser neutralizado. Estos sistemas que proponemos deben ser sometidos a una serie de procedimientos de minimización representados por tres fases de DM: relajación, equilibrio y muestreo, como se recomienda (Wang, 2020).
Igualmente debe ser considerada la incorporación en este tipo de modelos de reclasificación de funciones de puntuación variables asociadas a la actividad biológica, tales como pIC50, previos ajustes inherentes a la incompatibilidad conocida entre las funciones de puntuación y los valores de pIC50 (Huang y Zou, 2010). Especialmente, porque nosotros encontramos en análisis preliminares mediante el uso del algoritmo PASS (Prediction of Activity Spectra for Substances) (Lagunin y col., 2018) que la posibilidad de que muchos de nuestros compuestos estudiados pertenezcan a la subclase de compuestos activos es tan alta como la del Remdesivir. Lo que corresponde con las predicciones previas en términos de logIC50 (µM) que realizamos en diversas líneas celulares mediante el uso del servidor DiPCell (Kumar y col., 2014) y que muestran como algunos de nuestros compuestos pueden presentar una actividad inhibitoria teórica más favorable que Remdesivir (resultados no mostrados). Consideraciones que, de ser incorporadas en nuestro modelo en estudios posteriores, pero ajustadas después de un análisis profundo de todas las variables involucradas a la interacción ligando-proteína podrían predecir de manera más realista el comportamiento de estas moléculas en ensayos in vitro, y más aún si se busca una aplicación inmediata de este tipo de compuestos frente al COVID-19.
4. CONCLUSIONES
Todos los compuestos fitoquímicos probados son capaces de establecer interacciones termodinámicamente favorables con la RNA polimerasa viral según la predicción realizada por los algoritmos probados y las funciones de puntuación consideradas. El fitoquímico capaz de establecer el acoplamiento más favorable con la proteína viral fue la piperina, superando al control Remdesivir. La mitad de los fitoquímicos evaluados presentó acoplamientos más favorables que el antiviral comercial, así como una mejor bioacumulación teórica global. Las calificaciones derivadas de las funciones de puntuación se ven afectadas por las condiciones de los acoplamientos, especialmente por la solubilidad de los ligandos, la relación total de hidrofobicidad e índices de hidropatía de las cavidades. La eficacia de las funciones de puntuación, para una correcta clasificación de los acoplamientos para ligandos solubles con átomos o grupos químicos que puedan establecer enlaces tanto polares como no polares acoplados a cavidades hidrofóbicas, se puede ordenar de la siguiente manera: Rerank > MolDock > PLANTS > AutoDock Vina > DockT. Se propone el uso del algoritmo MolDock para cribados rápidos y al Rerank para reorganizar la clasificación de los acoplamientos cuando se trabaje con ligandos solubles, indistintamente de la polaridad, y que estén dirigidos a cavidades hidrofóbicas de la RNA polimerasa viral del SARS-CoV-2. Se necesita validar la reproducibilidad de estos resultados con un número mayor de ligandos y proteínas, así como incorporar nuevas variables asociadas a la dinámica molecular y actividad biológica para proponer el desarrollo y aplicación de un algoritmo de reclasificación basado en características hidrofóbicas de los complejos ligando-proteína y que funcione de manera genérica acoplado a las funciones de puntación ya descritas derivadas de los algoritmos de acoplamiento, para continuar con la optimización de la discriminación de los mejores fármacos durante la investigación y desarrollo de candidatos en las etapas in silico frente al COVID-19.