









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. INTRODUCCIÓN
Los pesticidas organoclorados (OCLs) son contaminantes orgánicos persistentes que han sido ampliamente utilizados desde 1950 para proteger los cultivos de las plagas (Li et al., 2014; Moon et al., 2009; Soceanu et al., 2012; Wu et al., 2014). La evidencia del peligro producido por ellos en el medio ambiente y la salud se ha demostrado en las últimas décadas (Florax et al., 2005; Pimentel, 2005), dada a la aplicación de técnicas analíticas de separación.
Para garantizar que los alimentos, piensos y agua sean inocuos y aptos para el consumo, las comisiones reguladoras internacionales han establecido límites máximos de residuos (LMRs) (Dömötörová & Matisová 2008; Xu et al., 2013). En Ecuador, muchas organizaciones agrícolas están preocupadas por el impacto de estos compuestos, ya que no existen mecanismos para controlar su uso adecuado (Crissman, et al., 1994; Donald C. et al., 2002; Hurtig, et al., 2003; Paz-y-Miño, et al., 2002), a pesar de los cambios citogenéticos demostrados en los agricultores tras la exposición a estos compuestos tóxicos. Inclusive si los pesticidas son aplicados con buenas prácticas agrícolas (BPA), pueden dejar residuos que pueden causar efectos nocivos para la salud y el medio ambiente, aún si se encuentran en concentraciones muy bajas (Masci, et al., 2014; Štěpán, et al., 2005).
Los análisis de OCLs pueden llevarse a cabo por cromatografía de gases acoplada con detector de captura de electrones (GC/ECD), ofreciendo una alta eficiencia de separación y una mayor selectividad (Goñi, et al., 2009; Rial-Otero, et al., 2007). Por otro lado, esta técnica carece de poder de identificación. Pese a que existen técnicas más rápidas como tándem MS, tiempo de vuelo o trampa MS de iones, estos requieren de materiales y equipos costosos (Peré-Trepat, et al., 2007) y las instituciones privadas e inclusive las organizaciones gubernamentales no lo poseen. La cromatografía de gases acoplada con espectrometría de masas (GC-MS) es la técnica de análisis más utilizada para el análisis de residuos de pesticidas (Alder, et al., 2006; Matisová & Dömötörová, 2003). Detecta los analitos de interés a través de la selección de iones específicos determinados con el monitoreo de iones seleccionados (LeDoux, 2011), proporcionando información selectiva y sin interferencia espectral (Cajka, et al., 2008; Hernández, et al., 2006).
Un método accesible para controlar la materia prima en Ecuador tiene que ser certificado, por consiguiente, el objetivo de este trabajo es desarrollar y validar un procedimiento analítico rápido, sensible y selectivo para la identificación y cuantificación residual de una mezcla multicomponente de OCLs que son comúnmente utilizados en la actividad agrícola ecuatoriana.
2. METODOLOGÍA
2.1 Químicos y estándares analíticos
La mezcla certificada multicomponente de pesticidas organoclorados MIX 13 del Dr. Ehrenstorfer MIX 13 Augsburg (Germany): Aldrin, cis-Clordana (alfa), trans-clordano (gama), oxyclordano, 2,4'-DDD, 4,4'-DDD, 2,4'-DDE, 4,4'-DDE, 2,4'-DDT, 4,4'-DDT, Dieldrin, Endrin, alfa-Endosulfan, beta-Endosulfan, alfa-HCH, beta-HCH, gamma-HCH, delta-HCH, epsilon-HCH, Heptacloro, Heptacloro-exo-epóxido (cis-, isómero B), Heptacloro-endo-epóxido (trans-, isómero A), Hexaclorobenceno, Isodrin, Metoxicloro, Mirex, 2,4,4'-Triclorobifenilo, 2,2',5,5'-Tetraclorbifenilo, 2,2',4,5,5'-Pentaclorobifenilo, 2,2',3,4,4',5'-Hexaclorobifenilo, 2,2',4,4',5,5'-Hexaclorobifenilo, 2,2',3,4,4',5,5'-Heptaclorobifenilo fueron analizados. Todas las disoluciones fueron almacenadas en la oscuridad a 4oC, protegidas de la luz y dejadas por 1 hora a temperatura ambiente antes de su uso.
2.2 Equipo
Un cromatógrafo de gases Agilent 6890N equipado con dos detectores diferentes, un espectrómetro de masa AGILENT 5973 y un detector de captura de electrones, además de un inyector automático AGILENT 7683. La separación de los picos fue analizada en dos columnas capilares con fase estacionaria diferente, una columna DB-5ms (0,25 mm x 30 m x 0,25 µm) de polímero fenil arileno equivalente a (5%- fenil)-metilpolisiloxano y una columna DB-35ms (0,25 mm x 30 m x 0,25 µm) equivalente a (35%- fenil)- metilpolisiloxano. Helio (99,999%) y nitrógeno (99,999%) fueron utilizados como gas portador y gas auxiliar respectivamente, ambos provenientes de la empresa AGA.
2.3 Condiciones del GC-MS
Las siguientes condiciones se evaluaron: Quince rampas de temperatura fueron probadas para el horno, tres temperaturas del inyector (230, 250 y 280°C), dos tiempos de saturación del solvente en el inyector (1 y 2 min), dos flujo de gas de arrastre (1 y 2 mL/min), tres flujo de purga (5 mL/min @ 1 min, 5 mL/min @ 0 min y 10 mL/min @ 1 min), tres modos de inyección (pulsed split 15 psi @ 2 min, 30 psi @ 2 min y una en modo splitless).
La influencia de cada factor se analizó por experimentación monofactor y las mejores condiciones instrumentales se seleccionaron en términos de resolución y abundancia relativa.
El detector de espectrometría de masa con impacto electrónico de 70 eV fue operado en modo SIM (monitoreo de ion(es) selectivo), el voltaje del multiplicador de electrones se fijó en 1480V. Las temperaturas de la fuente de iones y la línea de transferencia fueron ajustadas a 230 y 260°C, respectivamente. Cada compuesto fue identificado a través de la comparación con la biblioteca Wiley7n. El programa usado fue MSD ChemStation Build 26 Agosto 2003 de Copyright © Agilent Technologies 1989-2003.
Metodologías distintas fueron probadas en modo de barrido total SCAN y dos pruebas mediante el modo de detección de iones seleccionados SIM. El detector universal operado en modo full SCAN permitió la identificación y el análisis del espectro de masas, mientras con el detector específico operado en modo SIM se seleccionaron tres iones característicos de cada uno, para su análisis confirmatorio.
Una vez seleccionadas las condiciones, las muestras se inyectaron mediante el detector selectivo de captura de electrones con los siguientes parámetros: 250°C; Constant column + makeup flow; flujo combinado de 60 mL/min y nitrógeno como makeup gas.
2.4 Validación del método
El análisis de precisión se realizó mediante la determinación del coeficiente de variación y la linealidad de las curvas de calibración por el coeficiente de determinación. Para el espectrómetro de masas, seis diferentes concentraciones del pesticida MIX-13 fueron preparadas (0,05; 0,1; 0,25; 0,5; 0,75 y 1 mg ∙ L-1), por triplicado (repetibilidad) en tres días diferentes (reproducibilidad), mientras que para el detector de captura de electrones, se inyectaron cinco concentraciones (0,05; 0,25; 0,5; 0,75 y 1 mg ∙ L-1) con tres repeticiones de cada uno en un solo día.
Para la determinación de los límites de detección (LDD) y cuantificación (LDC), la concentración más baja que dio una señal diferente al del ruido se inyectó nueve veces en tres días diferentes, la señal media (Ys) y desviación estándar (Ss) se calcularon. La señal mínima detectable se calculó con la Ecuación (1):
El LDD se calculó a través de la Ecuación (2) lineal procedente de la curva de calibración:
El LDC se calculó usando la misma Ecuación (2), pero Y se calculó mediante la Ecuación (3):
3. RESULTADOS Y DISCUSIÓN
El proceso de desarrollo del método inició con la optimización de los parámetros instrumentales, enfocándose en tres elementos principales del sistema cromatográfico: El sistema de inyección, de separación y de detección.
En análisis de trazas como en la detección de pesticidas, el modo de inyección splitless es el más usado, dado al incremento de la sensibilidad (Chasteen, s. f.). Yang, et al. (2007) dentro de su estudio evaluaron el modo de inyección, siendo el modo pulsed splitless el que mostró mejor resultado. Tras comparar los modos de inyección, se determinó que el modo splitless ofrece una mayor abundancia de los picos cromatográficos que las dos pruebas realizadas en modo pulsed split, mas no existió efecto en la resolución de los picos cromatográficos. Por ejemplo el compuesto 2,4,4’- Triclorobifenilo en modo splitless tiene un área 25% mayor que las otras pruebas utilizando pulsed split. La resolución entre el 2,4,4’- Triclorobifenilo y delta-HCH fue 3,4, 3,3 y 3,5 usando splitless, pulsed split 15 psi y pulsed split 30 psi respectivamente.
Según Stashenko y Martínez (2011), la temperatura del inyector tiene que ser identificada para asegurar la volatilidad de todos los compuestos y la no degradación de los mismos. La temperatura se programó en un rango de 230 a 280℃ para impedir la ruptura de la mayoría de los compuestos termolábiles (Martínez Vidal, et al., 2000). Aunque la temperatura del inyector es útil para aumentar las áreas cromatográficas; en este caso, se demostró que no existió ninguna diferencia en la resolución y la abundancia de los picos. La temperatura seleccionada fue de 250°C. El tiempo de saturación del solvente en el inyector (1 y 2 min) tampoco mostraron diferencia, siendo 1 min el tiempo seleccionado.
Tras seleccionar el modo de inyección, se evaluaron tres pruebas del flujo de purga: 5 mL/min (1 min), 5 mL/min (0 min) y 10 mL/min (1 min). Los resultados mostraron que utilizando 5 mL/min (1 min) se exhibió mayor área cromatográfica, seguido por 5 mL/min (0 min). Para el caso del 2, 2´, 4, 4´, 5, 5´-hexaclorobifenilo, al usar el primer flujo se logró áreas dos y cuatro veces mayores que usando el segundo y tercer flujo.
Al analizar la diferencia entre el flujo de gas de arrastre (1 y 2 mL/min) no se encontró mayor diferencia basándonos en resolución cromatográfica, por lo que se seleccionó 1 mL/min, permitiendo un ahorro significativo en el consumo de gas.
Comparando las dos columnas, una columna DB-5ms y una columna DB-35ms, se determinó que la primera columna expone ligeramente una mayor abundancia, mejor resolución y además de lograr una mayor identificación en el número de compuestos. Con la columna DB-35 ms se logró determinar que nueve de los treinta y dos compuestos coeluyen: epsilon-HCH con 2,2',5,5'-tetraclorbifenilo y con aldrin; 2,4'–DDE con 2,2',4,5,5'-pentaclorobifenilo; 4,4'–DDD con 2,4'–DDT; 2,2',3,4,4',5'-hexaclorobifenilo con 4,4'–DDT. Con la columna DB-5 ms coeluyen ocho compuestos: oxi-clordano con heptacloro-exo-epóxido; cis-clordano con alfa-endosulfan; 4,4'–DDD con 2,4'–DDT; 2,2',3,4,4',5'-hexaclorobifenilo con 4,4'–DDT.
Con el fin de separar todos los OCLs, se probaron quince programas de temperatura del horno, centrándose en la eficiencia de la separación de los picos y tiempo de ejecución; siendo la programación de horno de la columna: 70°C (durante 2 min) → 120°C (25°C/min) → 220°C (8°C/min, durante 10 min) → 250°C (25°C/min, durante 7 min).
Las condiciones instrumentales empleadas en esta metodología permitieron la identificación de veinticuatro pesticidas organoclorados, pues ocho de los treinta y dos presentes en el PESTICIDE MIX-13 coeluyen (ver Figura 1). Para la separación de los compuestos que compartieron el mismo tiempo de elución, se realizaron dos pruebas mediante SIM con los iones cualificantes de cada compuesto. En la primera corrida se seleccionaron los iones de oxi-clordano (115, 187 y 149), cis-clordano (373, 375 y 377) y de 4,4'–DDT (235, 237 y 165) y para la segunda se trabajó con los iones de heptacloro-exo-epóxido (353, 355 y 351); alfa-endosulfan (241, 239 y 195); y, de 2,2',3,4,4',5'-hexaclorobifenilo (360, 362 y 290). La Figura 2 muestra los cromatogramas obtenidos con la metodología en las dos pruebas SIM, en la que se aprecian las abundancias de dichos compuestos.
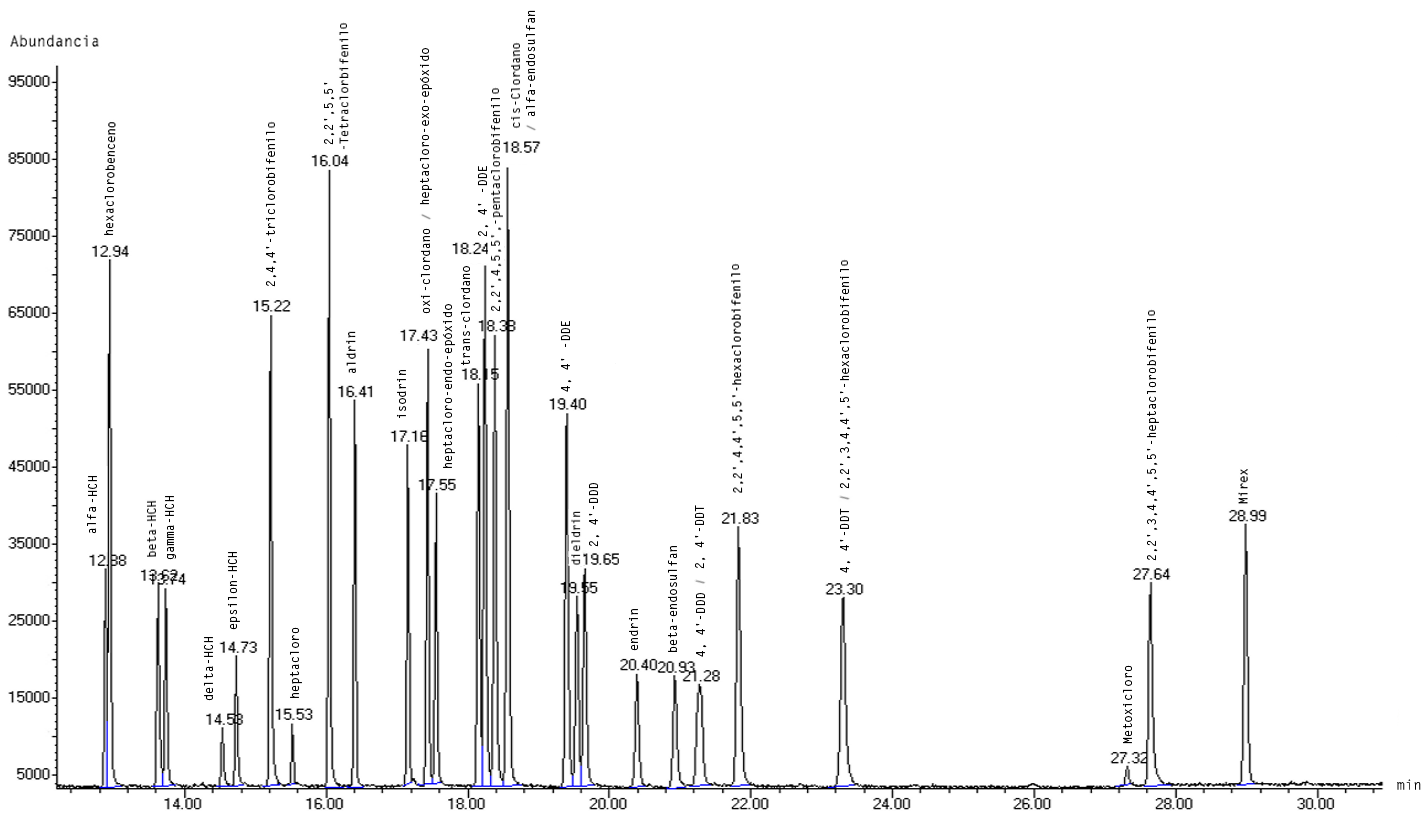
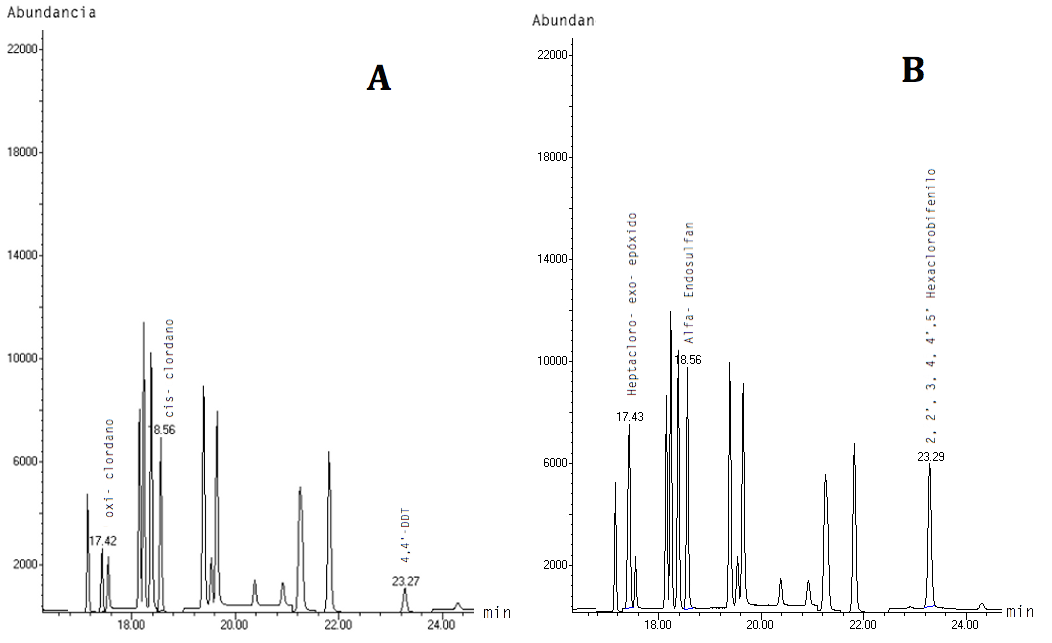
Figura 2. Grupos en modo SIM. A). Grupo 1: Clordano, cis- clordano y 4,4’-DDT B). Grupo 2: Heptacloro-exo-epóxido, alfa- endosulfan y 2, 2’, 3, 4, 4’ 5’ - Hexaclorobifenilo
Tabla 1. Tiempo de retención, coeficiente de determinación, pendiente e intersección conseguidos con las condiciones seleccionadas
| Pesticida | Detector | TR(min) | Coeficiente determinación | Pendiente (L/mg) | Intersección | ||||||
| Alpha-HCH | MS | 12,89 | 0,997 | ± | 0,001 | 55 | ± | 2 | -23 | ± | 5 |
| ECD | 13,98 | 0,999 | ± | 0,001 | 152 | ± | 3 | -6 | ± | 7 | |
| Hexaclorobenceno | MS | 12,94 | 0,999 | ± | 0,001 | 154 | ± | 4 | -13 | ± | 14 |
| ECD | 14,05 | 1,000 | ± | 0,000 | 104 | ± | 2 | 7 | ± | 5 | |
| beta-HCH | MS | 13,63 | 0,994 | ± | 0,003 | 53 | ± | 23 | 1 | ± | 20 |
| ECD | 14,70 | 0,998 | ± | 0,001 | 67 | ± | 1 | -13 | ± | 3 | |
| gamma-HCH | MS | 13,74 | 0,995 | ± | 0,002 | 59 | ± | 1 | -21 | ± | 10 |
| ECD | 14,84 | 0,997 | ± | 0,001 | 115 | ± | 2 | -44 | ± | 4 | |
| delta-HCH | MS | 14,53 | 0,991 | ± | 0,005 | 21 | ± | 1 | -26 | ± | 6 |
| ECD | 15,37 | 0,996 | ± | 0,002 | 11 | ± | 1 | 7 | ± | 2 | |
| epsilon- HCH | MS | 14,73 | 0,995 | ± | 0,003 | 36 | ± | 3 | -15 | ± | 7 |
| ECD | 15,61 | 0,997 | ± | 0,001 | 101 | ± | 2 | -31 | ± | 6 | |
| 2,4,4´-Triclorobifenilo | MS | 15,22 | 0,998 | ± | 0,001 | 142 | ± | 3 | -56 | ± | 15 |
| ECD | 15,82 | 0,998 | ± | 0,000 | 65 | ± | 1 | -10 | ± | 2 | |
| Heptacloro | MS | 15,53 | 0,999 | ± | 0,001 | 15 | ± | 1 | 2 | ± | 2 |
| ECD | 16,29 | 0,999 | ± | 0,001 | 34 | ± | 1 | 6 | ± | 3 | |
| 2, 2´,5,5´-Tetraclorobifenilo | MS | 16,05 | 0,999 | ± | 0,001 | 165 | ± | 4 | -48 | ± | 14 |
| ECD | 16,63 | 0,999 | ± | 0,000 | 55 | ± | 2 | -17 | ± | 4 | |
| Aldrin | MS | 16,41 | 0,998 | ± | 0,001 | 100 | ± | 3 | -40 | ± | 10 |
| ECD | 17,15 | 0,999 | ± | 0,001 | 16 | ± | 1 | 5 | ± | 3 | |
| Isodrin | MS | 17,16 | 0,998 | ± | 0,002 | 94 | ± | 4 | -35 | ± | 7 |
| ECD | 17,61 | 0,999 | ± | 0,001 | 152 | ± | 3 | 18 | ± | 5 | |
| Heptacloro-endo-epóxido (trans-, isómero A) | MS | 17,56 | 0,995 | ± | 0,003 | 98 | ± | 3 | -62 | ± | 16 |
| ECD | 18,84 | 0,999 | ± | 0,001 | 227 | ± | 4 | 3 | ± | 7 | |
| Trans-Clordano | MS | 18,15 | 0,998 | ± | 0,001 | 131 | ± | 5 | -54 | ± | 13 |
| ECD | 18,98 | 0,999 | ± | 0,001 | 138 | ± | 2 | 2 | ± | 3 | |
| 2, 4´-DDE | MS | 18,25 | 0,998 | ± | 0,001 | 186 | ± | 5 | -87 | ± | 18 |
| ECD | 19,72 | 0,999 | ± | 0,001 | 146 | ± | 3 | -5 | ± | 6 | |
| 2,2´,4,5,5´-Pentaclorobifenilo | MS | 18,39 | 0,999 | ± | 0,001 | 169 | ± | 3 | -59 | ± | 12 |
| ECD | 19,76 | 0,999 | ± | 0,000 | 81 | ± | 2 | 5 | ± | 4 | |
| 4,4'-DDE | MS | 19,40 | 0,996 | ± | 0,002 | 150 | ± | 5 | -84 | ± | 14 |
| ECD | 20,23 | 0,999 | ± | 0,001 | 257 | ± | 5 | 31 | ± | 11 | |
| Dieldrin | MS | 19,55 | 0,999 | ± | 0,001 | 69 | ± | 7 | -18 | ± | 5 |
| ECD | 21,21 | 0,999 | ± | 0,001 | 147 | ± | 3 | -1 | ± | 6 | |
| 2,4'-DDD | MS | 19,66 | 0,999 | ± | 0,001 | 69 | ± | 8 | -20 | ± | 5 |
| ECD | 21,42 | 0,999 | ± | 0,000 | 136 | ± | 3 | 13 | ± | 4 | |
| Endrin | MS | 20,39 | 0,995 | ± | 0,003 | 41 | ± | 10 | -1 | ± | 6 |
| ECD | 21,48 | 0,999 | ± | 0,000 | 102 | ± | 1 | 3 | ± | 3 | |
| beta-Endosulfan | MS | 20,93 | 0,994 | ± | 0,003 | 49 | ± | 7 | -20 | ± | 7 |
| ECD | 22,51 | 1,000 | ± | 0,000 | 56 | ± | 2 | -7 | ± | 7 | |
| 2,2',4,4',5,5'-Hexaclorobifenilo | MS | 21,83 | 0,997 | ± | 0,001 | 142 | ± | 5 | -64 | ± | 13 |
| ECD | 23,52 | 0,998 | ± | 0,000 | 157 | ± | 2 | -25 | ± | 4 | |
| Metoxicloro | MS | 27,32 | 0,995 | ± | 0,005 | 9 | ± | 1 | -2 | ± | 4 |
| ECD | 27,78 | 0,997 | ± | 0,003 | 5 | ± | 0 | 5 | ± | 2 | |
| 2,2',3,4,4',5,5'-Heptaclorobifenilo | MS | 27,65 | 0,993 | ± | 0,004 | 102 | ± | 6 | -91 | ± | 14 |
| ECD | 28,17 | 0,996 | ± | 0,001 | 30 | ± | 1 | -10 | ± | 4 | |
| Mirex | MS | 28,99 | 0,996 | ± | 0,002 | 116 | ± | 5 | -47 | ± | 9 |
| ECD | 29,29 | 0,998 | ± | 0,000 | 62 | ± | 1 | -1 | ± | 3 | |
| TR: Tiempo de Retención | |||||||||||
| Para MS: Pendiente x 10 000, Intersección x 1000 | |||||||||||
| Para ECD: Pendiente x 1E+08, Intersección x 1E+07 | |||||||||||
Tabla 2. Límites de detección y cuantificación para MS y máximos límites de residuos para alimentos y agua de consumo
| Pesticida | LMRs | Límite de Detección (MS) | Límite de Cuantificación (MS) | |||||
| Cereales | Agua | Alimentos | Agua | Alimentos | Agua | |||
| (ppm) | (ppb) | (ppm) | (ppb) | (ppm) | (ppb) | |||
| alfa-HCH | 0,2 c3 | - | 0,006 | 0,097 | 0,01 | 0,146 | ||
| Hexaclorobenceno | 0,01b | 1a | 0,005 | 0,069 | 0,008 | 0,113 | ||
| beta-HCH | 0,2 c3 | - | 0,008 | 0,113 | 0,019 | 0,284 | ||
| gamma-HCH | 0,01b | 0,2a | 0,007 | 0,104 | 0,012 | 0,176 | ||
| delta-HCH | 0,2 c3 | - | 0,021 | 0,319 | 0,028 | 0,415 | ||
| epsilon-HCH | 0,2 c3 | - | 0,01 | 0,144 | 0,013 | 0,195 | ||
| 2,4,4'-Triclorobifenilo | - | 0,5a2 | 0,006 | 0,086 | 0,008 | 0,125 | ||
| Heptacloro | 0,01b3 | 0,4a | 0,006 | 0,084 | 0,01 | 0,143 | ||
| 2,2',5,5'-Tetraclorobifenilo | - | 0,5a2 | 0,005 | 0,081 | 0,008 | 0,12 | ||
| Aldrin | 0,01b4 | 0,03c2 | 0,006 | 0,095 | 0,01 | 0,147 | ||
| Isodrin | - | - | 0,006 | 0,085 | 0,007 | 0,111 | ||
| oxy-Clordano | 0,2c2 | 0,009* | 0,132* | 0,016* | 0,237* | |||
| Heptacloro-exo-epóxido (cis-, isómero B) | 0,01b3 | 0,2a1 | 0,009* | 0,132* | 0,016* | 0,237* | ||
| Heptacloro-endo-epóxido (trans-, isómero A) | 0,01b3 | 0,2a1 | 0,008 | 0,119 | 0,012 | 0,181 | ||
| trans-Clordano (gamma) | 0,02c1 | 0,2c2 | 0,006 | 0,093 | 0,009 | 0,14 | ||
| 2, 4'-DDE | - | - | 0,006 | 0,088 | 0,009 | 0,128 | ||
| 2,2',4,5,5'-Pentaclorobifenilo | - | 0,5a2 | 0,006 | 0,085 | 0,009 | 0,129 | ||
| cis-Clordano (alfa) | 0,02c1 | 0,2c2 | 0,006* | 0,097* | 0,010* | 0,148* | ||
| alfa-Endosulfan | 0,05b2 | - | 0,006* | 0,097* | 0,010* | 0,148* | ||
| 4, 4'–DDE | 0,05b1 | 1c2 | 0,006 | 0,095 | 0,008 | 0,122 | ||
| Dieldrin | 0,01b4 | 0,03c2 | 0,006 | 0,09 | 0,01 | 0,146 | ||
| 2, 4'–DDD | - | - | 0,006 | 0,094 | 0,011 | 0,165 | ||
| Endrin | 0,01b | 2a | 0,008 | 0,117 | 0,014 | 0,206 | ||
| beta-Endosulfan | 0,05b2 | - | 0,007 | 0,11 | 0,01 | 0,149 | ||
| 4, 4'–DDD | 0,05b1 | 1c2 | 0,014* | 0,211* | 0,023* | 0,348* | ||
| 2, 4'-DDT | 0,05b1 | 1c2 | 0,014* | 0,211* | 0,023* | 0,348* | ||
| 2,2',4,4',5,5'-Hexaclorobifenilo | - | 0,5a2 | 0,006 | 0,093 | 0,008 | 0,12 | ||
| 4, 4'–DDT | 0,05 b1 | 1c2 | 0,013* | 0,189* | 0,020* | 0,305* | ||
| 2,2',3,4,4',5'-Hexaclorobifenilo | - | 0,5 a2 | 0,013* | 0,189* | 0,020* | 0,305* | ||
| Metoxicloro | 2c | 40 a | 0,024 | 0,355 | 0,037 | 0,549 | ||
| 2,2',3,4,4',5,5'-Heptaclorobifenilo | - | 0,5 a2 | 0,012 | 0,179 | 0,017 | 0,254 | ||
| Mirex | - | - | 0,006 | 0,095 | 0,008 | 0,126 | ||
| a La Agencia de Protección Medioambiental (EPA) establece máximo nivel de contaminantes para sustancias tóxicas en agua de consumo. Disponible en http://water.epa.gov/drink/contaminants/index.cfm#one | ||||||||
| a1 Valores referidos a heptacloro epóxido | ||||||||
| a2 Valores referidos a Bifenilos policlorados | ||||||||
| b LMRs establecidos por la Unión Europea. Disponible en http://ec.europa.eu/sanco_pesticides/public/?event=pesticide.residue .selection&language=EN | ||||||||
| b1 Sum of p,p´-DDT, o,p´-DDT, p-p´-DDE and p,p´-TDE (DDD) expressed as DDT | ||||||||
| b2 Suma de isómeros alfa- y beta- y endosulfan-sulfato expresados como endosulfan | ||||||||
| b3 Suma de heptacloro y heptacloro epóxido expresado como heptacloro | ||||||||
| b4 Aldrin y dieldrin combinados expresados como dieldrin | ||||||||
| c LMRs establecidos por la Fundación Japonesa de Investigación en Química de Alimentos. Disponible en http://www.m5.ws001.squarestart.ne.jp /foundation/search.html | ||||||||
| c1 LMRs para clordano están establecidos como la suma de los residuos de cis-clordano y trans-clordano en productos agrícolas | ||||||||
| c2 Valores fijados para agua mineral (agua mineral natural y aguas de consumo envasadas/embotelladas) | ||||||||
| c3 Suma de isómeros del BHC, a excepción de isómero gamma | ||||||||
| * Límites de detección y cuantificación están expresados como la suma de los compuestos que comparten el mismo tiempo de retención | ||||||||
Al co-eluir y poseer los mismos iones característicos (235, 237 y 165), el 4,4'–DDD y 2,4'–DDT no pudieron ser identificados mediante SIM, más recurriendo a un ajuste en la rampa de temperatura: 70°C (durante 2 min) → 190°C (25°C/min) → 235°C (8°C/min) → 250°C (30°C/min, durante 10 min),se logró una separación con 0,67 de resolución; en contraste, los compuestos restantes redujeron su abundancia e inclusive algunos pesticidas no fueron identificados.
La precisión entre ensayos para la mayoría de los compuestos es muy buena en los tres días de análisis, dado a que el coeficiente de variación se encuentra por debajo del 14%, lo cual nos indica la reproducibilidad del método. Únicamente para el caso del beta-HCH, 2,4'-DDD y endrin, este parámetro tiene valores superiores, por lo que se podría optar por utilizar un estándar interno.
La linealidad del método fue probada por ECD y MS mediante el ajuste lineal entre el área de los picos frente a la concentración correspondiente para generar la curva de calibración. La respuesta del detector MS fue lineal en el intervalo de 0,05 a 1 mg L-1, a excepción de beta-HCH con un intervalo de 0,05 a 0,75 mg ∙ L-1, delta-HCH desde 0,25 a 1 mg ∙ L-1, epsilon-HCH de 0,1-0,75 mg ∙ L-1, metoxicloro desde 0,25 a 1 mg ∙ L-1 y 2,2 ', 3,4,4', 5,5'-heptaclorobifenilo de 0,1 a 1 mg ∙ L-1. Para ECD, las concentraciones analizadas fueron 0,05; 0,25; 0,5; 0,75 y 1 mg ∙ L-1, todas ellas con un coeficiente de determinación superior a 0,99 (ver Tabla 1).
Este método puede utilizarse como análisis de control de calidad dado que la LDD y LDC fueron inferiores a los niveles máximos de residuos de alimentos y de agua establecidos por organismos reguladores (ver Tabla 2). Sólo para el aldrin y el dieldrin este método no sería apropiado para evaluar en agua de consumo.
4. CONCLUSIONES
La metodología desarrollada permitió la identificación de veinticuatro compuestos organoclorados en la misma serie cromatográfica a través del empleo de un detector de captura de electrones y un análisis confirmatorio por espectrometría de masas. En el caso de 4,4'-DDD y 2,4'-DDT, que coeluyen y poseen los mismos iones característicos, se realizó un ajuste en la rampa de temperatura, con lo cual se logró una resolución de 0,67.
La eficiencia del método fue demostrado por la obtención de un coeficiente de variación menor al 14% y un coeficiente de determinación mayor a 0,99; siendo este método una herramienta accesible para controlar los pesticidas en el Ecuador.