









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Vogt-Koyanagi-Harada (VKH) es una enfermedad sistémica inflamatoria poco frecuente de etiología autoinmune, caracterizada por un cuadro de uveítis granulomatosa posterior bilateral, asociado a síntomas neurológicos, auditivos y dermatológicos.
Este cuadro es explicado por un proceso de auto-inmunidad iniciada por linfocitos T dirigida contra antígenos de superficie de melanocitos, afectando a tejidos con alto contenido de estas células como es el caso de la retina, meninges, oído medio, piel y fanéreos. Se ha demostrado que este proceso de autoinmunidad está fuertemente asociado a factores de riesgo genéticos en población de piel pigmentada, se desconoce hasta el día de hoy un gatillante claro de esta enfermedad. Compromete de manera temprana al tejido meníngeo y vestibulococlear, presentándose como meningismo y tinnitus o con el antecedente de estos síntomas, a medida que la patología avanza compromete la retina, manifestándose como pérdida rápida de agudeza visual bilateral, ya en etapas crónicas afecta piel y fanéreos, presentando poliosis y vitíligo. El tratamiento de elección es la inmunomodulación mediante el uso de corticoides en altas dosis, con mejores resultados a largo plazo cuanto más temprano se inicie este tratamiento.
Esta enfermedad concierne tanto a oftalmología, neurología y otorrinolaringología, especialidades que podrían enfrentar la consulta de un paciente con enfermedad de Vogt-Koyanagi-Harada. A continuación, se presenta una revisión de la bibliografía disponible acerca de esta enfermedad.
La enfermedad de Vogt-Koyanagi-Harada fue inicialmente descrita en 1906 por Alfred Vogt, quien describió el caso de un paciente con uveítis anterior asociado a poliosis y vitíligo, al igual que Einosuke Harada en 1926 y Yoshizo Koyanagi en 1929. En el año 1955 se habla por primera vez de la enfermedad de Vogt-Koyanagi-Harada, nombre por el cual es conocida hasta el día de hoy.(1) Habiendo establecido una entidad nosológica independiente bajo 3 nombres, comienza el esfuerzo de la comunidad científica por caracterizar y entender de mejor manera esta enfermedad.
EPIDEMIOLOGÍA
La enfermedad de Vogt-Koyanagi-Harada corresponde a una enfermedad rara de base autoinmune que ocupa un lugar relevante entre las causas de uveítis posterior y panuveítis de etiología no infecciosa, junto a la enfermedad de Behcet y Sarcoidosis.(2) Posee una distribución mundial, con variaciones según ubicación geográfica y grupo étnico siendo más frecuente en personas de piel pigmentada, con una mayor predisposición descrita en poblaciones asiáticas, latinoamericanas y de oriente medio, en la población africana subsahariana la enfermedad de VKH es poco frecuente a pesar de corresponder a una etnia de piel pigmentada.(3) Al contrario, es una enfermedad rara en población caucásica de tez clara, con pocos o nulos casos reportados en algunos países europeos,(2) siendo Italia el país que reporta la proporción más alta.(4)(5) (Tabla 1)
Tabla 1 Prevalencia de la enfermedad de Vogt-Koyanagi-Harada respecto de otras causas de uveítis, en distintas regiones del mundo.
| Region/Pais | Porcentaje | Autor/Año |
|---|---|---|
| ASIA | ||
| China | 13.5% (2079/15373) | Yang et al, 2020 (6) |
| China | 15.9% (278/1752) | Yang et al, 2005 (7) |
| China | 20.6% (125/606) | Gao et al, 2016 (8) |
| Japón | 7% (267/3830) | Ohguro et al, 2009 (9) |
| Japón | 9.7% (120/1240) | Kitamei et al, 2009 (10) |
| India | 4.3% (55/1123) | Sabhapandit et al, 2016 (11) |
| India | 3% (57/1912) | Dogra et al, 2016 (12) |
| Singapur | 8.7% (109/1249) | Siak et al, 2016 (13) |
| Tailandia | 13.5% (102/758) | Sukavatcharin et al, 2016 (14) |
| Filipinas | 9.2% (55/595) | Abaño et al, 2017 (15) |
| LATINOAMÉRICA | ||
| Brasil | 7.5% (79/1053) | González et al, 2016 (16) |
| Chile | 17.2% (105/611) | Liberman et al, 2014 (17) |
| Argentina | 21.4% (73/341) | Hurtado et al, 2014 (18) |
| Colombia | 0.95% (25/2638) | Guayacán et al, 2017 (19) |
| MEDIO ORIENTE | ||
| Arabia Saudita | 22% (195/888) | Al Dhibi et al, 2016 (20) |
| Arabia Saudita | 19.6% (126/642) | Al Dhahri et al, 2014 (21) |
| Irán | 3.9% (21/544) | Soheilian et al, 2004 (22) |
| Turquía | 1.2 % (9/761) | Kazokoglu et al, 2008 (23) |
En China corresponde a la primera causa de uveítis no infecciosa con una prevalencia de 13.5% en un estudio reciente,(6)(7)(8) mientras que en Japón, corresponde a la segunda causa después de Sarcoidosis, (9)(10) con cifras similares en otros países de la región, a excepción de India. (11)(12)(13)(14)(15) En Latinoamérica destacan países como Brasil con prevalencias que oscilan entre 2.5% a 7.5% en publicaciones recientes, Chile y Argentina presentan las cifras más altas reportadas en la región, probablemente atribuible a un componente genético predisponente en común, (16)(17)(18) mientras que un estudio realizado en Colombia muestra la prevalencia más baja de la región. (19) En Medio Oriente, el país con mayor prevalencia es Arabia Saudita con una prevalencia para VKH que ha aumentado a 19% - 22% según estudios más recientes. (20)(21)(22)(23) Para mayor información sobre estudios epidemiológicos en regiones de alta prevalencia revise la Tabla I.
Afecta principalmente a personas entre la segunda y quinta década de la vida, con casos poco frecuentes en mayores de 65 años y menores de 16 años, edades en que el diagnóstico es dificultoso debido a su baja sospecha. La prevalencia de VKH en población pediátrica se estima entre 3% a 13% según la literatura, con una mayoría de casos reportados en Medio Oriente, (24) se describen cuadros clínicos más agresivos y de peor pronóstico visual respecto a otros grupos etarios, lo que depende principalmente del estadio clínico al momento del diagnóstico y el inicio de tratamiento oportuno. (25) La proporción de pacientes adultos mayores con VKH respecto a otras causas de uveítis ronda el 15% de casos en distintas series, con mayor incidencia de complicaciones oculares, requerimiento de dosis mayores de corticosteroides, con agudeza visual residual tras el tratamiento relativamente bien preservada que no difiere respecto a pacientes más jóvenes. (26)
La enfermedad demuestra predisposición por el sexo femenino, siendo la diferencia proporcional entre mujeres y hombres menos marcada en población asiática, e incluso mostrando leve predominancia del sexo masculino en algunas publicaciones de la región. (3)(4)
FISIOPATOLOGÍA
Si bien la etiopatogenia de la enfermedad de Vogt-Koyanagi-Harada no ha sido determinada por completo, se cuenta con evidencia clínica y experimental que sugiere un mecanismo de autoinmunidad mediado por linfocitos T contra antígenos de superficie de melanocitos, lo que se condice con el compromiso de los tejidos con este tipo de células, como la retina, meninges, oído medio, piel y fanéreos.(27)(28)
El genotipo del complejo mayor de histocompatibilidad tipo II HLA-DRB1*04 es el factor de susceptibilidad más importante, específicamente los subtipos HLA-DRB1*04.05 y HLA-DRB1*04.10, el que ha presentado una alta asociación con un OR promedio de 10.3 (95% IC: 5.56-19.11) y 6.5 (95% IC: 3.23-13.18) respectivamente según un metaanálisis,(29) se encuentra presente en prácticamente todo paciente que desarrolla la enfermedad en población Japonesa. (30) Estos sub alelos del genotipo HLA determinan reactividad exaltada de linfocitos T CD4+, con respuesta tipo Th1 contra proteínas de la familia de las tirosinasas (tyrosinase related protein 1 y 2, tyrosinase, gp100), grupo que se encuentra involucrado en la producción de melanina y expresada exclusivamente en melanocitos.(27)(28) Estos genotipos también son posibles de encontrar en otras enfermedades autoinmunes como la sarcoidosis o la enfermedad de Behcet, e incluso en personas sanas, sin embargo, la reactividad inmunitaria exaltada contra melanocitos se observa exclusivamente en pacientes con enfermedad de VKH. (28) En otras poblaciones distintas a la japonesa, la proporción de pacientes con el gen de susceptibilidad HLA-DRB1*04.05 y su fuerza de asociación con enfermedad de VKH es menor, esto podría ser explicado por la presencia de otros genotipos HLA o no HLA menos conocidas que determinen autoinmunidad contra otras proteínas de melanocitos no del todo estudiadas, como HLA-DQ, Interleukinas, antígeno 4 de linfocito T citotóxico, cuya expresión depende de factores étnicos y ambientales, (31) sin embargo, el genotipo HLA-DRB1*04 y sus subtipos persisten como el mayor factor de susceptibilidad para la enfermedad incluso en poblaciones con baja frecuencia de este genotipo. (32)(33)
El factor desencadenante de la enfermedad en pacientes genéticamente susceptibles es desconocido. Se ha propuesto que la inducción de autoinmunidad contra melanocitos podría ser gatillada tras infección viral, específicamente por Citomegalovirus (CMV) o virus Epstein-Barr (VEB), mediante un mecanismo de mimetismo molecular con proteínas superficiales de melanocito. Sugita et. al. demuestra una coincidencia molecular de 6 aminoácidos entre el antígeno de tirosinasa y la secuencia de Glicoproteína H de CMV (CMV-egH), además de una reactividad inmunitaria exaltada por parte de linfocitos T de pacientes con enfermedad de VKH tras la exposición a CMV-egH y Tirosinasa. (28)(34)
Existen reportes de casos que informan el inicio de enfermedad de VKH temporalmente cercana entre contactos, lo que sugiere la posibilidad de un gatillante infeccioso, (35) así como la detección de DNA de VEB mediante reacción de polimerasa en cadena en cuerpo vítreo de un paciente con enfermedad de VKH, (36) y el reporte de asociación entre enfermedad de VKH e infección concomitante por Virus Influenza A, (37) información a considerar con cautela, son necesarios mayores estudios para esclarecer la asociación entre el inicio de enfermedad de VKH e infección viral.
A la fecha, solo se cuenta con unos cuantos reportes de casos relacionando la enfermedad de VKH con la vacunación. Respecto de la vacunación COVID-19, se ha encontrado un riesgo relativo similar comparado a otro tipo de vacunas. (38)(39)
MANIFESTACIONES CLÍNICAS
La enfermedad de Vogt-Koyanagi-Harada tiene una evolución clínica natural bien establecida con variaciones marcadas entre los distintos estados. Se han descrito 4 fases de la enfermedad. (40)
1.- Fase prodrómica
Periodo inicial, con una duración de 3-5 días, caracterizada por síntomas inespecíficos como malestar generalizado, fiebre, debilidad, síntomas flu-like, fotofobia, náuseas, dolor orbitario, y síntomas neurológicos como cefalea, tinnitus, rigidez nucal y dificultad auditiva, hallazgos neurológicos presentes respectivamente en 49%, 36%, 33% y 32% de los casos en un estudio. En exámenes diagnósticos se puede evidenciar pleocitosis en líquido cefalorraquídeo hasta en 77% de los casos de VKH que puede persistir hasta por 8 semanas. (41)(42)
2.- Fase uveítica
Con una duración de varias semanas, el paciente consulta por pérdida de agudeza visual bilateral, síntoma cardinal en este periodo. Un 30% de los pacientes presentan pérdida de agudeza visual monocular, es esperable un desfase de 3 a 10 días hasta comprometer el ojo contralateral. Al examen ocular se pone en evidencia un proceso inflamatorio granulomatoso en forma de uveítis posterior en etapas tempranas, lo que se traduce en engrosamiento coroideo, desprendimiento de retina seroso, hiperemia/edema de disco óptico presente respectivamente en 50%, 50% y 47% de los casos en un estudio. El proceso inflamatorio puede extenderse desde la úvea posterior a la cámara anterior, constituyendo una panuveítis en etapas avanzadas. (41)(42)
3.- Fase de convalescencia
Fase de meses a años de duración, predominan signos secundarios a un proceso de despigmentación de piel y fanéreos con vitíligo simétrico, alopecia, poliosis de cejas, pestañas y cabello. A nivel ocular ocurre despigmentación coroidal, dando paso al ̈signo de Sugiura ̈, despigmentación o vitíligo perilímbico (signo precoz de despigmentación) y “sunset glow fundus”, despigmentación coroidal con nervio óptico pálido, lo que da un aspecto rojo-anaranjado al examen de retina como muestra la figura 1. (41) En estadios crónicos, se describe una incidencia de 31% para poliosis, 22% vitíligo y 19% de alopecia, respecto a manifestaciones oculares se informa una prevalencia de 65% para sunset glow fundus y de 6% para el signo de Sugiura, este último descrito casi exclusivamente en población japonesa, ambos signos se presentan en ninguno de los pacientes en etapas agudas, siendo altamente sugerentes de VKH crónico. (42)
4.- Fase crónica-recurrente
Etapa crónica, como resultado de una terapia inmunosupresora inadecuada o sub-óptima con actividad inflamatoria activa, se caracteriza por episodios agudos de uveítis anterior recurrente, con el consecuente desarrollo de complicaciones como glaucoma, cataratas, fibrosis subretinal, neovascularización coroidal y finalmente, atrofia coriorretinal. (40)(41)
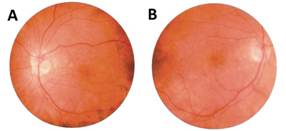
DIAGNOSTICO
El diagnóstico de la enfermedad de VKH es clínico, se basa en la confluencia de síntomas y signos sugerentes, asociado a exámenes complementarios de apoyo. Se han realizado varios esfuerzos para conformar los mejores criterios diagnósticos, ninguno de ellos es infalible ni tiene la aprobación unánime de los especialistas. Los criterios del Comité internacional de nomenclatura “Revised Diagnostic Criteria for VKH Disease” (RDC), (43) han sido utilizados desde su publicación en 2001 (Tabla II). Estos criterios clasifican la enfermedad en 3 categorías posibles:
-Enfermedad de VKH Completa: Compromiso ocular bilateral asociado a hallazgos extraoculares neurológicos/auditivos y dermatológicos.
-Enfermedad de VKH Incompleta: Compromiso ocular bilateral asociado a uno de los dos hallazgos extraoculares, neurológicos o dermatológicos.
-Enfermedad de VKH Probable: Cuadro clínico que presenta únicamente compromiso ocular. (Tabla 2)
Tabla 2 Criterios diagnósticos de la Enfermedad de Vogt-Koyanagi-Harada. Adaptado de Read et al, American Journal of Ophthalmology, 2001.(43)
| 1. Ausencia de antecedente traumático ocular penetrante o cirugía que preceda el cuadro de uveítis. |
| 2. Ausencia de evidencia clínica o de laboratorio que sugieran una enfermedad ocular distinta. |
| 3. Compromiso ocular bilateral. A.- Manifestaciones tempranas - Evidencia de coroiditis difusa que se manifiesta como fluido subretinal en áreas focalizadas o desprendimiento de retina seroso buloso. - Si la presencia de coroiditis es dudosa, debe presentar áreas focales retinales con retraso de perfusión, áreas placoides de hiperfluorescencia, tinción de nervio óptico por angiografía con fluoresceína asociado a engrosamiento coroideo difuso, sin evidencia de escleritis posterior en ultrasonografía. B.- Manifestaciones tardías - Historia previa de manifestaciones descritas. - Despigmentación ocular: Sunset glow fundus o signo de Sugiura. - Otros signos oculares: Lesiones coriorretinianas despigmentadas numulares, acumulación/migración del epitelio retinal pigmentario, uveítis anterior recurrente o crónica |
| 4. Hallazgos o antecedentes neurológicos/auditivos: Meningismo, tinnitus o pleocitosis en líquido cefalorraquídeo. |
| 5. Hallazgos dermatológicos (No precede al compromiso ocular ni del sistema nervioso central): Alopecia, poliosis o vitíligo. |
Debido a la similitud clínica y de laboratorio entre la enfermedad de VKH y oftalmía simpática, es necesaria la exclusión estricta de antecedentes de trauma o cirugía ocular, otras enfermedades con manifestaciones oculares similares a la enfermedad de VKH como sarcoidosis, uveítis sifilítica, uveítis tuberculosa dentro de las que deben ser descartadas. El compromiso ocular debe ser bilateral, en etapas tempranas, se debe confirmar un proceso de coroiditis difusa, de ser dudosa se requiere estudio de retina con lámpara de hendidura asociado a angiografía con fluoresceína/verde indocianina, ultrasonografía y/o tomografía para demostrar compromiso retiniano (Figura 2). En etapas tardías (meses a años), el signo de sunset glow fundus (despigmentación coroidal) o signo de Sugiura (vitíligo de limbo esclerocorneal), son altamente específicos para la enfermedad de VKH (Figura 1). Entre los síntomas neurológicos se incluyen cefalea, náuseas, fiebre, rigidez de nuca o tinnitus, de no presentar sintomatología neurológica, es necesario buscar activamente pleocitosis en líquido cefalorraquídeo o evidencia imagenológica que demuestre compromiso meníngeo (Figura 3). Las manifestaciones dermatológicas ocurren tras meses a años de evolución y no deben preceder al compromiso ocular. (43)
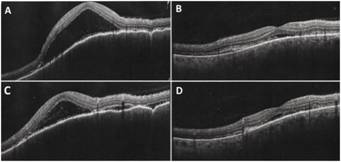
Figura 2 Tomografía de coherencia óptica (OCT), ojo derecho (A-B) y ojo izquierdo (C-D). Se observa desprendimiento exudativo de retina con separación entre epitelio pigmentario y retina neurosensorial. En la imagen C se logra apreciar la segmentación característica del desprendimiento de retina seroso.
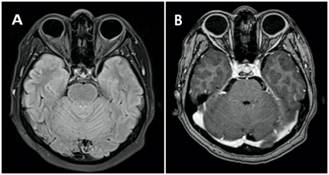
Figura 3 Resonancia nuclear magnética pretratamiento, imagen axial en Flair con supresión de la grasa (A). Imagen axial en T1 contrastada con gadolinio (B). Se observa engrosamiento coroideo bilateral con realce tras la administración del contraste e hiperintensidad de señal en flair retrobulbar.
Comparado con otros criterios diagnósticos, como los criterios de Sugiura ampliamente utilizados en Japón, y los criterios de la American Uveitis Society utilizados previamente en occidente, los criterios RDC integran al diagnóstico de VKH a pacientes con manifestaciones clínicas tanto agudas como crónicas con mayor detalle y descripción de los síntomas y signos, sin determinar tiempo de evolución de síntomas. Ha demostrado alta sensibilidad y especificidad diagnóstica, incorpora por primera vez como elemento diagnóstico exámenes imagenológicos como la angiografía con fluoresceína y ultrasonografía.(44)(45)
Cabe destacar que la separación en grupos completa, incompleta y probable no se correlaciona con la severidad de la enfermedad ni se asocia a presencia de alelo HLA-DRB1*0405, con una baja proporción de pacientes que clasifiquen en el grupo de VKH completa (10-15%) en distintas series, lo que podría ser explicado por la consulta en estadio temprano de la enfermedad e influencia del tratamiento corticoesteroideo temprano sobre la evolución natural de la enfermedad, entre otros factores, (44)(45) incluso algunos autores argumentan que la enfermedad de VKH probable, sin manifestaciones extraoculares (También llamado en algunas publicaciones VKH Ocular), debiese ser considerado VKH genuino. Es claro que el grupo VKH probable necesita mayor desarrollo en futuras clasificaciones.
Nuevos esfuerzos de conformar criterios diagnósticos han sido realizados recientemente por Yang et al en población China, (46) con enfoque en clasificar la enfermedad respecto a etapa de evolución en temprana/tardía según las manifestaciones clínicas y estudios complementarios, lo que se correlaciona de mejor manera con la evolución, pronóstico y tratamiento del cuadro clínico, lo cual tendría mayor utilidad en la práctica médica. (47) Demostró ser más sensible y poseer un valor predictivo negativo significativamente superior respecto a los criterios RDC. Es necesario corroborar la validez de estos criterios diagnósticos en pacientes de poblaciones distintas a la estudiada. (46)
TRATAMIENTO Y PRONÓSTICO
El objetivo del tratamiento de la enfermedad de VKH corresponde a limitar la progresión y acabar de manera temprana y agresiva con el proceso inflamatorio en contra de melanocitos.
El tratamiento se basa en el uso de corticoides sistémicos en altas dosis, se inicia habitualmente con 1-2 mg/Kg de peso al día de Prednisona o dosis equivalente, (48) ya sea vía oral o endovenosa sin diferencia significativa en resultados terapéuticos o incidencia de complicaciones iatrogénicas entre ambas opciones. (49) Se aconseja asociar inmuno moduladores no esteroideos en pacientes con mala respuesta inicial a glucocorticoides o con predictores de mala respuesta a tratamiento corticoesteroideo (Principalmente pacientes con comorbilidades y fondo de ojo con signos de despigmentación al momento de presentación), de preferencia utilizar tacrolimus y ciclosporina A aprovechando su acción citostática frente a linfocitos T. (48)(50) El uso de fármacos biológicos ha sido utilizado exitosamente en casos de uveítis no infecciosa, sin embargo, la evidencia acerca de su uso especifico en enfermedad de VKH es escasa. Adalimumab corresponde a un agente anti-factor de necrosis tumoral (Anti-TNF) con utilidad en el tratamiento de VKH en estado crónico-recurrente, ha demostrado mejorar significativamente la agudeza visual a la vez que permite disminuir dosis de corticoesteroides e inmunosupresores. Su uso es seguro, con baja incidencia de efectos adversos, principalmente infecciones oportunistas o condiciones autoinmunes como psoriasis. (51)(52)(53) Los implantes de corticoesteroides intravítreos no son utilizados actualmente en ningún tipo de uveítis, considerando su asociación a complicaciones severas como glaucoma y cataratas. La cirugía intraocular se reserva únicamente para la corrección de complicaciones, no se utiliza como tratamiento primario. (54)
El tratamiento debe durar un tiempo mínimo de 6 meses, con disminución paulatina de dosis hasta suspender, el tratamiento de menor duración se asocia a recurrencia y peor pronóstico visual. (55) Se debe tener en cuenta que el tratamiento corticoesteroideo subóptimo puede controlar clínicamente la enfermedad, sin necesariamente resolver el proceso inflamatorio subyacente ni las lesiones a nivel retinal, contribuyendo a la progresión subclínica del cuadro y al fracaso de la terapia a largo plazo. Por esta razón, la evolución y efectividad de la terapia debiese idealmente ser monitorizada mediante angiografía con verde indocianina, capaz de detectar compromiso inflamatorio de vasculatura coroidea. (56)(57) El inicio de un tratamiento adecuado en etapas tempranas de la enfermedad ha demostrado un mejor pronóstico visual a largo plazo, menores tasas de avance hacia cronificación y menor tasa de recurrencias. (58) La definición de terapia de inicio temprana difiere entre distintas publicaciones, debido a que no existe consenso en el concepto de enfermedad aguda de inicio temprano entre distintos autores y no forma parte de los criterios diagnósticos utilizados actualmente. (47) Se acepta como tratamiento temprano a aquel iniciado dentro de 2 a 3 semanas desde el comienzo de los síntomas, ventana de oportunidad en la que se demuestra mayor efectividad del tratamiento según la literatura. (58)
Las complicaciones de la enfermedad de VKH son principalmente oculares, ocurren entre el 40-50% de los casos e incluye la aparición de cataratas, glaucoma, membranas neovasculares subretinianas y fibrosis subretinal. (59)(60)
Varios aspectos influyen sobre el pronóstico visual del paciente. Se asocia a buen pronóstico clínico el inicio de un tratamiento corticoesteroideo adecuado y oportuno, uso de inmunomoduladores, agudeza visual inicial mejor que 20/200 y VKH en estadio agudo inicial al momento de la consulta, en cambio, se asocia a mal pronóstico clínico la aparición de complicaciones visuales (principalmente cataratas y sunset glow fundus), VKH crónico-recurrente y la presencia de manifestaciones extraoculares. La edad <18 años ha demostrado resultados contradictorios entre distintas publicaciones, clásicamente se ha considerado un factor de mal pronóstico visual, sin embargo, se acepta que la edad >18 años es factor de riesgo para el desarrollo de complicaciones. (59)(60)
CONCLUSIÓN
Se ha intentado resaltar la variabilidad clínica propia de la enfermedad de VKH y el desafío diagnóstico que conlleva. Al ser una entidad infrecuente y multifacética, resulta especialmente difícil su diagnóstico.
Aún no ha sido posible determinar la etiopatogenia de esta enfermedad por completo, se ha conseguido evidencia clínica y experimental que sugieren mecanismos de autoinmunidad mediada por linfocitos T contra antígenos de melanocitos en población genéticamente susceptible, lo que explica sus manifestaciones clínicas, se desconoce hasta el día de hoy un gatillante claro de este fenómeno autoinmune, se sospecha de un mecanismo de mimetismo molecular, sin embargo, es necesario recabar mayor evidencia que apoye esta hipótesis.
El diagnóstico es eminentemente clínico asociado a exámenes de apoyo imagenológico y de laboratorio, son necesarios futuros esfuerzos en mejorar los criterios diagnósticos de la enfermedad de VKH, con especial énfasis en la detección de estadios tempranos en vista a determinar un tratamiento oportuno en la práctica clínica y llegar a un consenso diagnóstico en la práctica investigativa, facilitando el mayor conocimiento y estudio de esta rara patología.
El tratamiento se basa en el uso de corticoides sistémicos en altas dosis de manera temprana por periodos prolongados, asociado a medicamentos inmunomoduladores o biológicos, para luego disminuir la dosis paulatinamente hasta suspender en el transcurso de meses. La complicación más común e importante corresponde a la formación de cataratas y glaucoma, los factores pronósticos más importantes que determinan el resultado visual a largo plazo para el paciente son principalmente el momento de inicio de terapia y la agudeza visual al momento de la consulta.