









 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
A monoclonal gammopathy (MG), also known as paraproteinemia, is an accumulation of the same immunoglobulin type due to an abnormal clonal proliferation of B-cells.(1)(2) They are usually divided into heavy chain (IgM, IgG, or IgA) and light chain (kappa or lambda) paraproteinemias. The B-cell single clone proliferation can sometimes be secondary to a hematological malignancy.(2) When a hematological malignancy is not present, the monoclonal gammopathy is considered to have an undetermined significance (MGUS). MGUS occur in 3.2% of persons over the age of 50 and 5.3% of persons over the age of 70.(3)
Polyneuropathies are prevalent in the elderly population, present in 4.4% of persons in the eighth decade and 13.2% of persons in the ninth decade of life.(4) Both monoclonal gammopathies and polyneuropathies are more common in men.(3)(4)(5) Monoclonal gammopathies, either of undetermined significance or those associated with a hematological malignancy, frequently co-exist with polyneuropathies.(5)(6) Thus, the American Academy of Neurology guidelines recommend searching for a paraproteinemia with serum protein electrophoresis or immunofixation in patients with a polyneuropathy.(7) Based on the type of monoclonal gammopathy and/or polyneuropathy, this coexistence may have a common pathophysiology or else be due to a mere coincidence. Understanding the difference has obvious implications in the diagnostic work-up and subsequent treatment of these patients. The purpose of this manuscript is to review the features that will allow clinicians to correctly determine a causal association between a monoclonal gammopathy and polyneuropathy, as well as to summarize the diagnostic work-up and treatment of these conditions.
Monoclonal Gammopathy of Undetermined Significance
When a MG is detected either with serum protein electrophoresis or immunofixation, the first step is always to determine its significance, i.e., is it reflective of a hematological malignancy or is it part of a (relatively) benign process? Criteria have been developed to define a paraproteinemia as a MGUS, based on the amount of clonal burden(1)(5):
1.Serum monoclonal protein level of <3gr/dl.
2.Bone marrow biopsy showing less than 10% of plasma cells.
3.Urine in 24 hours showing less than 500 mg of monoclonal protein.
4.Absence of hypercalcemia, renal failure, anemia, or bone pain (CRAB).
Identifying a monoclonal gammopathy in a serum protein electrophoresis or immunofixation should prompt a referral to hematology to determine its significance. From a neurological perspective, it helps to divide the heavy chain MGUS into those that are IgM vs. non-IgM-related, since the former have a well-established association with polyneuropathy, whereas for the latter a co-existence between polyneuropathy and true MGUS is more likely to be coincidental (unless the monoclonal gammopathy is part of a hematological malignancy (see POEMS below]).(5) This division also has implications from a hematological perspective, as IgM MGUS are unlikely to progress to a multiple myeloma, but do have a risk of progressing to Waldenström macroglobulinemia;(8) conversely, non-IgM MGUS, have a significant risk of progression to multiple myeloma.(5)(9) In some cases, the monoclonal expansion is of a light chain only, either kappa or lambda; in these cases, there is a risk of progression to light-chain myeloma.(5) In all types of either heavy or light-chain MGUS, there is risk of an association with amyloidosis, thus it should always be considered in the differential.(1)(5)
IgM Monoclonal Gammopathies and Polyneuropathies
IgM Monoclonal Gammopathy of Undetermined Significance
IgM MGUS-associated polyneuropathy
Outside of the context of hematological malignancies, paraprotein of the IgM subtype is the only one that has been definitely associated with polyneuropathy. In this condition, intramyelinic deposits have been observed in electron microscopy as widenings of myelin.(1) On occasions, the specific IgM paraprotein is an anti-myelin associated glycoprotein (MAG) antibody, which is found in 50% of the patients presenting with this type of polyneuropathy.(1)(5) Katz and colleagues(10) divided a cohort of 53 patients with acquired demyelinating polyneuropathy into those with proximal and distal weakness with distal sensory loss (typical chronic inflammatory demyelinating polyradiculoneuropathy [CIDP] phenotype), and those with only distal weakness and sensory symptoms. In the latter group, the frequency of an IgM MGUS was 66%, compared with only 22% the typical CIDP phenotype group.(10) Patients with the distal phenotype and IgM MGUS had poor response to conventional CIDP treatment. This phenotype has been labeled distal acquired demyelinating symmetric polyneuropathy (DADS). Among those with the IgM MGUS and DADS phenotype, 67% were found to have positive anti-MAG antibodies in serum, and thus this type of neuropathy is also often referred to as anti-MAG polyneuropathy.(5) Still, the presence or absence of the anti-MAG antibody in DADS does not change the clinical presentation, response to treatment and prognosis of these patients.(10)(11) The presence of IgM MGUS or even anti-MAG does not always correlate with a DADS phenotype, as a proportion of these patients have a phenotype consisting of typical CIDP. Patients with the DADS phenotype have prominent distal sensory symptoms, often accompanied by sensory ataxia and tremor, with little to no weakness; if weakness is present, it is usually mild and affects only distal feet muscles.(5) In electrodiagnostic studies, there is a length-dependent pattern, with distal demyelination that presents as prolongation of the distal motor latencies and absent sensory responses.(12) Some authors have reported that the use of the terminal latency index, a measure that specifically assesses the distal velocity of the nerve, is helpful in distinguishing DADS from patients with hereditary demyelinating polyneuropathies,(13) although it did not help to distinguish between DADS and CIDP.(10) It must be noted that in the latest CIDP guidelines published in 2021 by the European Academy of Neurology/Peripheral Nerve Society, the recommended term for DADS phenotype is “distal CIDP”.(14) Despite the naming similarity, the recommendation regarding distal CIDP/DADS is for it to be considered as a separate entity from “typical” CIDP when it is accompanied by IgM MG.(14)
As mentioned above, patients with DADS phenotype and IgM MGUS do not usually respond satisfactorily to conventional CIDP therapy such as corticosteroids or intravenous immunoglobulin (IVIg).(5)(10) The most recent meta-analysis showed that there may be a small, although unclear if clinically significant benefit for IVIg, while rituximab seems to be more promising although stronger evidence is lacking.(15) Lack of homogeneity among patients is a frequent setback of these trials. Certain factors such as subacute course and proximal weakness (indicative of CIDP and not DADS phenotype) predicted a favorable response to rituximab among all patients with anti-MAG seropositivity.(16) More recently, genetic analysis has shown that around 50-60% of patients with IgM MGUS-related polyneuropathy have a somatic point mutation in the myeloid differentiation factor 88 gene, which is present in almost all patients with Waldenström macroglobulinemia (WM), for which ibrutinib, a tyrosine kinase inhibitor, is often effective.(17) This has opened the avenue for the use of this and other WM drugs in IgM MGUS-related polyneuropathy, and further research is ongoing.(18)
Chronic Ataxic Neuropathy with Ophthalmoplegia, M-protein, agglutination, and disialosyl antibodies: CANOMAD
In rare cases, the monoclonal protein of the IgM subtype reacts with the disialosyl epitopes found in the GD1b, GD3, GT1b, and GQ1b gangliosides, which presents as a distinct syndrome of chronic sensory ataxia with ophthalmoparesis, usually referred to as CANOMAD based on its clinical and serological features.(5)(19) Half the patients have cold agglutinins.(19) This disorder can have a relapsing and/or a progressive course, with predominantly proprioceptive loss although motor weakness can also be found.(19) Almost all patients have diplopia from oculomotor dysfunction.(19) It resembles Miller Fisher syndrome, and shares similar mechanisms,(20) but CANOMAD is by definition a chronic disorder.
To our knowledge, there are no ongoing clinical trials targeting CANOMAD treatment. Based on case reports and case series, the recommended treatment is IVIg, which may be followed by rituximab.(5)(19)(21)(22)
IgM Monoclonal Gammopathy associated with a hematological malignancy
Waldenström Macroglobulinemia
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma with an IgM paraproteinemia.(8)(23) Patients have symptoms related to cancer infiltration, such as pancytopenia, fever, weight loss, organomegaly, Bing-Neel syndrome, and/or symptoms related to the monoclonal protein accumulation, such as hyperviscosity or polyneuropathy.(8) The WM-related polyneuropathy is phenotypically similar to IgM-related polyneuropathy, and thus anti-MAG antibodies are often detected.(8)(18) In some cases, the phenotype resembles that of typical CIDP with proximal weakness.(5)(8) Treatment should be targeted against the lymphoma.
Non-IgM Monoclonal Gammopathies and Polyneuropathies
Non-IgM MGUS
A well-established association between non-IgM (i.e., IgA or IgG) MGUS and polyneuropathies has not been demonstrated.(5) Thus, when a patient with an unexplained polyneuropathy is found to have IgA or IgG MGUS, the possibility that the two findings are coincidental is likely.(1)(6) A key factor to consider here is that some non-IgM MG that arise as a result of an underlying malignancy do have well-defined associations with a polyneuropathy, as it will be discussed below. Thus, investigations should be made to determine that the non-IgM MG is in fact an MGUS before discarding an association with the patient’s polyneuropathy.
Non-IgM MG related to hematological malignancies
The POEMS syndrome is a paraneoplastic syndrome due to an underlying clonal plasma cell dyscrasia. The acronym refers to some of the features associated with the syndrome including Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal plasma cell disorder, and Skin changes.(24)
The clinical presentation is variable. The vast majority of patients present with polyneuropathy (96-100% of the patients).(25)(26) The neuropathy has subacute onset over months and is characterized by a symmetrical and ascending length-dependent deficits. It is typically sensorimotor and painful. The severity of the neuropathy can range from mild to moderate, to about a third of the patients being wheelchair or bedbound at the time of diagnosis. (26)
Organomegaly is present in 45-85% of the patients, most commonly involving the liver and spleen. (25) Patients can also develop endocrine dysfunction, in 67-84% of the cases. This can be manifested as gonadal, adrenal, thyroid or glucose metabolism dysfunction. (25)(27) Extravascular volume overload is common, and patients can develop peripheral edema, pleural or pericardial effusions, or even anasarca. The majority of patients have osteosclerotic lesions. (25) Skin changes are common, present in up to 89% of the patients, most commonly characterized by skin hyperpigmentation, but other features may include hemangiomas or telangiectasias, hypertrichosis, whitening or clubbing of the nails, acrocyanosis or thickening of the skin. (25)
Other features associated with POEMS include papilledema, present in about half of the patients, (28)(29) and increased predisposition for arterial or venous thrombosis (up to a third of patients). (25)(26)(30) Pulmonary manifestations include pulmonary hypertension, restrictive lung disease, and impaired diffusing capacity of the lung for carbon dioxide. (31)
Laboratory investigation in patients with suspected POEMS should include the evaluation for monoclonal plasma cell proliferation, which are Lambda light chain in 95% of the cases. (1)(25)(32) Vascular endothelial growth factor (VEGF) elevation is a classic feature and thought to correlate with disease activity. (18) VEGF cut-off of 771pg/ml in serum has been found to have 100% sensitivity and 92% specificity for POEMS. (33) Most recently, the N-terminal pro-peptide of type I collagen has been described as a diagnostic marker of POEMS. (34) Thrombocytosis is seen in more than half of the patients with POEMS, and erythrocytosis is present in 12-19%.(25)
Electrophysiological studies are consistent with a sensorimotor symmetrical distal neuropathy. Nerve conduction studies show conduction velocity slowing in intermediate nerve segments, suggesting that demyelination occurs predominantly in the nerve trunk. Conduction block is rare, as opposed to CIDP. In contrast to CIDP, the axonal loss on POEMS is more prominent, predominantly seen in the lower extremities. (26)(35) Demyelinating changes are more often found in the upper extremities. (1)
Frequently misdiagnosed with CIDP, (1)(26) the median time to diagnosis is of almost 1 year. Making a correct diagnosis is crucial to reduce morbidity, costs associated with delayed or erroneous diagnosis and therefore treatment. POEMS should be considered in any patient with an acquired demyelinating neuropathy and should prompt checking for a monoclonal gammopathy and VEGF levels. (18)
Diagnostic criteria have been proposed. (25) Diagnosis requires the presence of a polyneuropathy and the identification of a monoclonal plasma cell proliferative disorder, plus the presence of at least one major and one minor criteria. Major criteria include the presence of sclerotic bone lesions, elevation of VEGF, and Castleman disease (angiofollicular lymph node hyperplasia, a variant of POEMS that occurs without evidence of a clonal plasma cell disorder). Minor criteria are organomegaly, endocrine abnormalities, skin changes, extravascular volume overload, thrombocytosis/polycythemia, and papilledema. Once suspected, investigations should start with a skeletal survey or a whole-body CT. (36)
The pathogenesis of this disease is not well understood. Different mechanisms of peripheral neuropathy have been proposed, including endothelial injury caused by over-expression of VEGF in the nerves of patients with POEMS, leading to abnormal activation of endothelial cells. (37)
Treatment requires a multidisciplinary management approach. Therapy usually leads to favorable outcomes, and the survival in patients with POEMS is typically superior to those of patients with classical multiple myeloma. A detailed discussion of different treatment options is outside the scope of this review. For patients without disseminated bone marrow involvement (one to three isolated bone lesions), radiation alone is recommended. Systemic chemotherapy, with or without peripheral blood stem cell transplant, is preferred in patients with POEMS syndrome with widespread osteosclerotic lesions or bone marrow involvement. Neurological improvement can be delayed, usually seen 6 months after the systemic therapy is completed. (25)
-Multiple Myeloma
-Multiple myeloma (MM) is a hematological malignancy of abnormal clonal plasma cells in the bone marrow. (9) The polyneuropathy observed in patients with MM is usually due to chemotherapy. (5)(9) Pre-treatment, about 5-20% of patients do have a polyneuropathy, which is thought to be related to amyloid deposition rather than the MM itself. (1)(5) Electrodiagnostically, the pattern is that of a length-dependent, sensory and motor polyneuropathy. Due to the association with amyloid, these patients often have carpal tunnel syndrome and autonomic dysfunction (see below for amyloid-related polyneuropathy). (5) Treatment of the polyneuropathy consists of targeting the malignancy. An important consideration is that the standard first-line therapy includes the proteasome inhibitor bortezomib, which can cause polyneuropathy in up to 33% of cases. (9) See Figure 1. (Figure 1)
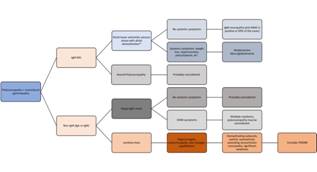
Figure 1 Algorithmic approach to patients with polyneuropathy and a heavy chain paraproteinemia. (MG: monoclonal gammopathy; anti-MAG: anti-myelin associated glycoprotein; CRAB: hypercalcemia, renal failure, anemia, and bone pain; POEMS: polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes; *: the phenotype in this case may also be that of a chronic inflammatory demyelinating polyradiculoneuropathy or CIDP).
Light-Chain MGUS and Polyneuropathies
Although all types of heavy and light-chain MG can be associated with amyloidosis, the MG is almost always light-chain type. More specifically, it tends to be of the lambda more than kappa light chain. (5) In light chain amyloidosis (AL), the light chains deposit in B-pleated sheets in different tissues. (18) It involves the nerves in 20-35% of the cases. (38) Contrary to the aforementioned MG-polyneuropathies, in AL the small fibers are the ones predominantly affected, which presents clinically as dysautonomia and painful dysesthesias. (2)(5) Often, AL does affect the axons of large-fibers as well causing length-dependent weakness. (5) On electrodiagnostic testing, a length-dependent sensory and motor axonal polyneuropathy is usually found, although if the AL is confined to the small fibers, electrodiagnostic testing may be normal. (5) Certain clues help the clinician suspect that a patient with polyneuropathy may have AL include the presence of bilateral carpal tunnel syndrome, cardiac arrhythmias, nephrotic syndrome, and unexplained weight loss. (18) Given its rarity, heterogeneity, and sometimes oligosymptomatic presentation, AL often has significant diagnostic delay. (38) Early suspicion of AL can lead to prompt diagnosis, as the combination of serum and urine immunofixation plus serum free light chains has a sensitivity of 100% of detecting the amyloidogenic chain. (39) The gold standard remains a biopsy, usually of the abdominal fat pad. (18)(38) Nerve biopsy has a lower sensitivity of 85%.(40) Skin biopsy can also help detect amyloid deposits. (41) There are also genetic causes of amyloidosis, of which the most common type is due to mutations in the transthyretin gene (TTR). (38) In these TTR-related cases there is no paraproteinemia, and the damage is due to a variant protein. It is only mentioned here because the polyneuropathy of TTR-amyloidosis is similar to that of AL amyloidosis, and it thus must be considered in the differential. (38) There have been significant advancements in the treatment of TTR-amyloidosis which are beyond the scope of this review. (38) Treatment of AL is targeted against the plasma cell clone, usually with chemotherapy that is similar to that used in MM, or in selected patients with autologous stem cell transplantation. (5)(28)
Other Considerations
Neurolymphomatosis
Rarely, a lymphoma may infiltrate the peripheral nerves directly. Neurolymphomatosis occurs in less than 0.5% of non-Hodgkin lymphomas, and it is even rarer in other lymphoma subtypes. (23) The presentation is varied, ranging from a mononeuropathy multiplex pattern which presents as asymmetric and sequential involvement of different nerves across time, to polyradiculopathy or cranial neuropathies. (18)(23) It should be suspected in patients with lymphoma and a paraproteinemia who develop one of the mentioned phenotypes and have not been exposes to neurotoxic chemotherapeutic agents. Cerebrospinal fluid examination is often abnormal, showing pleocytosis, hyperproteinorrachia, and hypoglycorrhachia, although cytology does not have great sensitivity. (23) The gold standard is a nerve biopsy, but recent studies show that nerve-dedicated magnetic resonance imaging and positron emission tomography-computed tomography scans are helpful in providing a diagnosis and/or selecting the nerve for biopsy. (18)(23) As is the case for other malignancy-related neuropathies, treatment is focused against the malignancy (Table 1).
Table 1 Summary of the types of monoclonal gammopathies that have a strong association with polyneuropathies. (DADS: distal acquired demyelinating symmetric polyneuropathy; CIDP: chronic inflammatory demyelinating polyradiculoneuropathy; anti-MAG: anti-myelin associated glycoprotein; CANOMAD: chronic ataxic neuropathy, ophthalmoplegia, M-protein, agglutination, and disialosyl antibodies; POEMS: polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes; VEGF: vascular endothelial growth factor.)
| Monoclonal gammopathy | Neuropathy phenotype | Systemic symptoms/other clinical manifestations | Laboratory/radiology findings | Management | |
|---|---|---|---|---|---|
| IgM Neuropathy | IgM | DADS, or less likely CIDP phenotype. | None, unless part of Waldenström. | Significantly prolonged distal motor latencies. 50% have positive anti-MAG antibodies. | Rituximab (inconclusive evidence). |
| CANOMAD | IgM | Ophthalmoplegia, sensory ataxia. | None. | GD1b, GDR, GT1b, and/or GQ1B antibodies. 50% have cold agglutinins. | IVIg followed by rituximab (weak evidence). |
| POEMS | IgA or IgG, usually with lambda light chain. | Demyelinating polyneuropathy, with proximal and distal motor and sensory impairment, pain, and progressive course. | Organomegaly, cardiac and/or pleural effusions, endocrine abnormalities, skin changes. | Elevated VEGF (> 771 pg/ml in serum). Thrombocytosis in 50% of the patients. Osteosclerotic lesion(s) in skeletal survey or whole-body CT scan. | Treating the underlying hematological malignancy: radiation, chemotherapy, stem cell transplantation |
| Light-chain Amyloidosis | Light chain, usually lambda (can be any type of MG). | Dysautonomia, painful dysesthesias. May also present as length-dependent weakness. | Cardiomyopathy, nephrotic syndrome, weight loss, carpal tunnel syndrome. | Nerve conduction studies may be normal. Biopsy of abdominal fat pad is the gold standard. | Treating the underlying plasma cell clone: chemotherapy or stem cell transplantation |
| Neurolymphomatosis | Any type | Asymmetric, sequential mononeuropathies | B symptoms. | Pleocytosis, low glucose, and elevated protein in CSF. MRI/PET scan show nerve abnormalities. | Treating the lymphoma. |
Conclusions
Making a diagnosis can be challenging in some of these patients, but it is crucial to prevent misdiagnosis, reduce morbidity and costs associated with delayed or erroneous diagnosis. Work-up for patients with polyneuropathy should include serum protein electrophoresis, immunofixation, and serum-free light chains. (36) Obtaining a good clinical history and physical examination will dictate further testing, such as radiographic assessment of bones, VEGF measurement, and analysis of bone marrow biopsy (see Figure 1 and Table 1). This extensive work-up should always be guided by the type and distribution of the symptoms, as well as by the electrodiagnostic findings. Gait ataxia due to large-fiber sensory loss distally in the lower extremities with little to no weakness, along with distal demyelination (prolonged distal latencies) in nerve conduction studies is consistent with a DADS phenotype which is the typical phenotype for an IgM MG. Proximal and distal, upper, and lower extremity weakness and sensory symptoms with either progressive or relapsing nature along with features of acquired demyelination (conduction blocks plus severe slowing of the conduction velocity) describe a CIDP phenotype, which may also be the presenting phenotype in IgM MG. On the other hand, autonomic dysfunction, bilateral carpal tunnel syndrome, and loss of pain and temperature sensation accompanied by systemic symptoms (cardiomyopathy, nephrotic syndrome, weight loss) strongly suggests amyloidosis which when caused by a paraproteinemia, it is almost always of the light chain type. Lastly, a patient with a sequential and often painful development of asymmetric wrist drop followed by foot drop and/or cranial neuropathies, in the context of a paraproteinemia or history of hematologic cancer should prompt the clinical for suspicion of neurolymphomatosis. In summary, recognizing the phenotypical presentations of polyneuropathies in the context of monoclonal gammopathies helps to accurately determine the significance of the association of these pathologies. Prompt recognition of these syndromes often leads to a specific diagnosis and an appropriate treatment course.