









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La meningioangiomatosis (MA) es una lesión del desarrollo, benigna e infrecuente de la leptomenínge y la corteza cerebral, asociada a epilepsia farmacorresistente (EFR). (1)(2)
Se reconocen dos categorías de MA, la esporádica y la asociada a neurofibromatosis tipo 2 (NF2). (1)(3) La mayoría de los casos se presenta de manera esporádica (75-80%) y el 25% asociada a NF2. (4) La variante esporádica se caracteriza por ser una lesión solitaria, que afecta a niños, adolescentes y adultos jóvenes con crisis intratables a los fármacos antiepilépticos (FAE). Presentan síntomas neurológicos inespecíficos como la cefalea o algún grado de déficit neurológico progresivo focal. (5) En la mayoría de los casos no se ha demostrado la mutación en el gen NF2. Por su parte, en la MA asociada a NF2, el rango de edad es variable (desde los 11 meses hasta los 73 años). (6) Los pacientes suelen ser asintomáticos, y en ellos se demuestra la presencia de mutaciones en el gen NF2. Es frecuentemente multifocal y resulta diagnosticada en los estudios postmortem. (4)
La asociación de MA con displasia cortical focal (DCF) ha sido escasamente publicada. (2)(6)(7)(8)(9) En este trabajo, presentamos el caso de una adolescente con EFR del lóbulo temporal derecho y diagnóstico histológico de DCF tipo IIIc (MA predominantemente vascular asociada a DCF).
Caso clínico
Se trata de una adolescente de 15 años y manualidad diestra. Sin antecedentes pre y perinatales, ausencia de crisis febriles y de trauma craneoencefálico. No se refieren antecedentes patológicos familiares. A los 5 años presenta la primera crisis (vómito, palidez y trastorno de la consciencia). Es ingresada y al alta médica le plantean el diagnóstico de crisis no epiléptica de posible naturaleza vasovagal. A los 6 meses del primer evento presentó un episodio de pérdida de la consciencia y movimientos en el hemicuerpo izquierdo. Se diagnostica epilepsia focal sin etiología precisada e indican tratamiento con carbamazepina. Evolutivamente aparecen otras crisis, que se inician con sensación epigástrica, presentando arresto de la actividad segundos después. Los eventos duran 1-2 minutos y se presentan tanto de día como de noche. Muy ocasionalmente tuvo crisis focales sin trastorno de consciencia y sin evolución ulterior. Recibió tratamiento psiquiátrico desde los 13 años por diagnóstico de crisis no epilépticas de naturaleza psicógenas. Las crisis eran diarias con una frecuencia de 5 /día. Estudió hasta 9no grado con dificultad.
Con estos antecedentes se realiza evaluación prequirúrgica protocolizada según el programa de cirugía de epilepsia del Centro Internacional de Restauración Neurológica. (10) La analítica sanguínea así como la exploración física general fueron normales. El examen neurológico demostró disminución de la atención, concentración y de la memoria a corto y largo plazo, así como un pensamiento lento.
Las imágenes de Resonancia Magnética (IRM) mostraron una lesión hiperintensa en T2 y FLAIR que produjo aumento de volumen difuso del hipocampo y región parahipocampal derecha, con presencia de pequeños vasos intracorticales. Se aprecia además adecuada diferenciación entre sustancia gris y blanca aunque con disminución de la sustancia blanca temporal derecha, lo que se hace evidente al compararla con la región temporal izquierda (Figura 1). En el estudio de T1 con gadolinio los vasos descritos realzaron con el contraste (ligera captación).
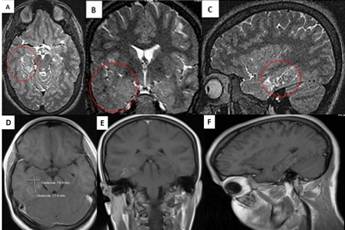
Figura 1 Imágenes de resonancia magnética en secuencia T2. A corte axial, B coronal, C sagital. Lesión levemente hiperintensa con engrosamiento y aumento de volumen difuso en región hipocampal y parahipocampal derecha. Los círculos rojos en A y C muestran pequeños vasos malformativos en región temporo mesial derecha. La imagen A y B evidencia disminución de la sustancia blanca temporal derecha y aumento del grosor cortical en región temporal lateral ipsilateral. La diferencia entre los lóbulos temporales homólogos se puede apreciar claramente al comparar el temporal derecho con el izquierdo. En la parte inferior de la figura, se muestran los cortes axial (D), coronal (E) y sagital (F) en secuencia T1 contrastados, donde se aprecia una ligera captación del contraste por los finos vasos malformativos en la región temporomesial derecha.
La evaluación prequirúrgica concluyó con el diagnóstico de crisis epilépticas focales sin alteración de la consciencia que evolucionan a crisis con trastorno de la consciencia. Se plantea su etiología estructural por posible lesión tumoral en hipocampo y parahipocampo derecho con componente vascular dependiente de la leptomenínge de la base craneal de la fosa media.
Previa discusión colectiva en el grupo de cirugía de epilepsia y con consentimiento informado, la paciente fue intervenida quirúrgicamente. Se le realizó lobectomía temporal derecha, con resección neocortical guiada mediante electrocorticografía (ECoG) intraoperatoria (Figura 2) y aspiración de estructuras mesiales (amígdala e hipocampo). Macroscópicamente la lesión fue dura, acartonada y muy vascularizada (vasos friables y sangrantes), extendiéndose desde el giro T3 hasta el giro parahipocampal y adherida a la duramadre del piso de la fosa media.
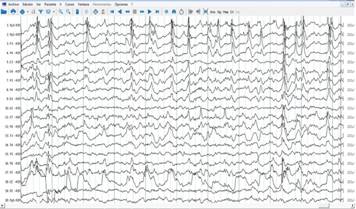
Figura 2 Segmento de Electrocorticograma intraoperatorio pre resección realizado con electrodos subdurales AdTech (malla 20 contactos). Nótese actividad puntas repetitivas con predominio de amplitud y frecuencia de descarga en el giro temporal inferior (T3). Amplitud registro 200 MicroV. Los contactos 1-5 registran giro temporal inferior T3, 6-9 T2 ,11-14 T2, 16-18 T1.
En el laboratorio de Anatomía Patológica se recibieron 6 fragmentos de tejido, todos fijados en formol tamponado al 10%. Del total, 4 fueron rotulados como neocorteza, el mayor de 3 x 1.5 x 1 cm. A los cortes se apreció un punteado hemorrágico focal y buena delimitación entre la sustancia gris y blanca. Los 2 fragmentos restantes fueron rotulados como tumor, el mayor de 1.5 cm con área de color pardo oscuro, forma alargada y consistencia firme. Las secciones histológicas de la corteza cerebral mostraron dislaminación de sus capas con disposición radial y tangencial, neuronas mal orientadas, algunas de ellas dispuestas verticalmente formando microcolumnas perpendicular a la superficie meníngea así como cambios espongióticos focales (fig. 3 E y F). El tejido señalado como tumor mostró un incremento marcado del número de vasos sanguíneos de diámetro variable con paredes engrosadas y hialinizadas y ausencia de células meningoteliales a su alrededor (fig. 3 A); focalmente se apreció la protrusión de estos hacia la corteza cerebral subyacente (fig. 3 B y C). En algunos vasos sanguíneos se observó positividad perivascular al antígeno de membrana epitelial (EMA) así como marcada gliosis del tejido cerebral interpuesto entre los vasos con la proteína ácida gliofibrilar (GFAP) (fig. 3 D). Se concluyó como DCF tipo IIIc (MA predominantemente vascular asociada a DCF). (Figura 3)
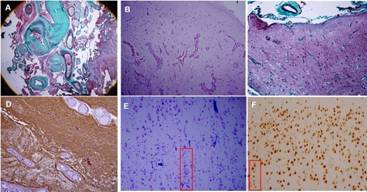
Figura 3 A. Meningioangiomatosis vascular caracterizada por numerosos vasos sanguíneos de diámetros variables, marcada hialinización de sus paredes y ausencia de células meningoteliales alrededor de los vasos (Tricrómica de Masson, 4x). B y C. Corteza cerebral subyacente a la lesión con protrusión de numerosos vasos sanguíneos congestivos (B - Hematoxilina-Eosina, 10x; C - Tricrómica de Masson, 10x). D. Tejido cerebral rarefacto cercano a la lesión con marcada gliosis y algunos vasos con paredes hialinizadas (GFAP, 20x). E. Corteza cerebral adyacente a la meningioangiomatosis. Se aprecia dislaminación de las capas III y IV con formación de microcolumnas perpendicular a la superficie meníngea (rectángulo), (Klüver-Barrera, 20x). F. Laminación cortical anormal de las capas II y III. El rectángulo evidencia 8 neuronas dispuestas verticalmente (microcolumna), (NeuN, 20x).
La paciente evolucionó satisfactoriamente y fue dada de alta. En los controles médicos realizados se mantuvo libre de crisis (IA en la escala de Engel) con 4 años de evolución postquirúrgica y sin FAE.
Discusión
La MA fue descrita en 1915 por Bassoe y Nuzum (11) al estudiar el cerebro de un adolescente de 15 años y diagnóstico de NF2. Años después, Worster-Drought et al. (12) publicaron un caso similar y fue nombrada como MA.
La patogénesis de la lesión es controvertida y confusa. Se plantean varias hipótesis: 1.- representa una malformación vascular cortical no caracterizada con proliferación de células meningoteliales; 2.- lesión hamartomatosa originada de los vasos leptomeníngeos y de la corteza cerebral con cambios degenerativos; 3.- invasión directa de un meningioma leptomeníngeo al parénquima cerebral adyacente a través de los espacios de Virchow-Robin y 4.- alteraciones genéticas en la región del gen NF2 (fuerte asociación con NF2). (4)(13)(14)
Tanto la MA esporádica como la asociada a NF2 muestran una predilección por el sexo masculino (13)(15)(16), aunque otros autores no refieren un predominio en cuanto al género. (17) La lesión comúnmente se localiza en la corteza cerebral. En los enfermos con MA esporádica el lóbulo temporal es el sitio más frecuente, similar al caso actual. En la MA asociada a NF2 el lóbulo frontal es la localización más común. Se han informado localizaciones infrecuentes como el cuerpo calloso, tercer ventrículo, tálamo, tallo cerebral y cerebelo. (3)(15)
La MA aparece hipodensa con realce del contraste y calcificación variable en la tomografía axial computarizada (TAC) en aproximadamente el 90% de los casos. (16) Los hallazgos en las IRM son inespecíficos y sugieren una malformación vascular (angioma cavernoso) o un tumor cerebral (meningioma y glioma de bajo grado entre otros). La lesión comúnmente es hipointensa en T1 e hiperintensa en T2.(16) Sun et al.(18), encuentran 3 casos de MA con patrones imagenológicos diferentes (sólido y quístico); los diagnósticos preoperatorios fueron de meningioma, hemangioblastoma y ganglioglioma. La presencia de una lesión calcificada en la TAC con anillo hipointenso en las IRM ponderadas en T2 son útiles para sugerir el diagnóstico de MA. (13)(15)(16) En nuestro caso, a pesar de la descripción imagenológica detallada, no se valoró la posibilidad de MA.
Macroscópicamente, la lesión es usualmente firme y bien delimitada del parénquima cerebral (18), aspecto similar al caso que presentamos. Al microscopio óptico se aprecia la presencia de vasos sanguíneos pequeños, proliferación perivascular de células fusiformes, hialinización así como fibrosis y calcificación. (4)(19) Histológicamente se puede clasificar en: - predominantemente celular, compuesta por vasos de paredes finas rodeados por células meningoteliales (celularidad de moderada a alta) que remeda un meningioma o - predominantemente vascular, con vasos sanguíneos de paredes gruesas, hialinizadas y calcificados con escasa proliferación celular perivascular que impresiona una malformación vascular. (15) El presente caso se incluye en esta última variedad. El perfil inmunohistoquímico ofrece positividad de las células fusiformes con la vimentina y detección variable con el CD34 y con el EMA. (15)
La MA puede asociarse con otras lesiones, tales como malformación arteriovenosa, oligodendroglioma, meningioma, quiste y DCF entre otros. (2)(6)(7)(8)(9)(20)(21)
La DCF es un tipo de malformación del desarrollo cortical (MDC) y causa frecuente de EFR. Histológicamente, la DCF se divide en 3 tipos. La tipo I (aislada) se caracteriza por una dislaminación cortical, radial (Ia), tangencial (Ib) y radial-tangencial (Ic). La variante II (aislada) se identifica por una laminación anormal de la corteza y la presencia de neuronas dismórficas (IIa) y células balonadas (IIb). La DCF tipo III se asocia a una lesión principal, IIIa (esclerosis hipocampal), IIIb (tumor glial o glioneuronal), IIIc (malformación vascular) y IIId (lesiones adquiridas durante la infancia como encefalitis y daño isquémico entre otras). (22) La asociación de MA y DCF está incluida en la variante tipo IIIc.
En niños con diagnóstico de EFR sometidos a cirugía, las MDC son el principal hallazgo histopatológico (39.3%). Dentro de este grupo, la DCF fue la causa más frecuente con el 70.6% de los casos. En la población adulta, las MDC ocupan el tercer lugar (19.8%). (23)
Se desconoce la etiología de la DCF. No obstante estudios genéticos y de secuenciación en las muestras quirúrgicas han evidenciado mutaciones en la vía mTOR (blanco de la rapamicina en mamíferos) en aproximadamente el 60% de los casos con DCF. (24) En el subtipo IIb se ha demostrado un incremento de la ruta de señalización mTOR (25), siendo negativo para las MDC ligeras y la DCF tipo I. (24)
En la actualidad se reconocen otras formas de MDC, entre las que se mencionan las MDC ligeras asociadas a hiperplasia oligodendroglial (MOGHE, de sus siglas en inglés) y la DCF tipo II de la parte inferior del surco que se localiza fundamentalmente en el lóbulo frontal. (26) El desarrollo de las neuroimágenes y los estudios genéticos han permitido ampliar el diagnóstico de los subtipos de DCF, lo que favorece una clasificación clínico-patológica-molecular, tal es el caso de la DCF tipo IIa con IRM positiva y mutación en la línea germinal DEPDC5 o de la DCF tipo IIId con IRM positiva y negativo para un amplio panel genético (mTOR, AKT1, AKT3, DEPDC5, TSC1, TSC2, PIK3CA, PIK3R2, NPRL2, NPRL3, PTEN, BRAF y SLC35A2). (26)
En una extensa revisión de la literatura, Roux et al. (9) refieren 175 pacientes con MA (incluyendo su caso). Del total, 109 (62.3%) presentaban EFR y en solo 14 enfermos (8%) se diagnosticó DCF. Dos artículos publicados en la década del 90 sobre MA han referido alteraciones en la neocorteza cercana a la lesión. Whiting et al. (27) describen un trastorno de la citoarquitectura cortical con neuronas morfológicamente anormales. De los 7 casos publicados por Wiebe et al. (15), uno presentó numerosas neuronas displásicas en la corteza adyacente a la lesión. Ambos trabajos hacen una descripción de los hallazgos microscópicos sin acuñar el término de DCF. Diferentes autores reseñan que la MA y la DCF son lesiones diferentes, pero comparten una fisiopatología común. Al ser la DCF un tipo de MDC, su asociación con MA apoya la etiología del desarrollo en la variante esporádica. (2)(4)(7)(9)(14)
La tabla 1 recoge los 15 casos publicados hasta la fecha de MA y DCF así como el presente caso. De los 16 pacientes, 12 corresponden al sexo masculino con un promedio de edad de 14.6 años en el momento del diagnóstico. (Tabla 1)
Tabla 1 Casos reportados en la literatura con diagnóstico de meningioangiomatosis y displasia cortical focal.
| Referencias Total pacientes | Caso (s) con Epilepsia intratable | Número / Edad / Género de casos con meningioangiomatosis y displasia cortical focal | Fármacos antiepilépticos | Evolución clínica postquirúrgica. Utilización fármacos antiepilépticos |
| Kim et al., 2009 6 9 | 8 | 1 / 23 / M | NM | LC NM |
| Batra and Prayson, 2013 7 1 | 1 | 1 / 23 / M | Carbamazepina | LC con FAE |
| Mukae et al., 2014 8 2 | 2 | 2 / 17 / M / 16 / M | NM | Ambos LC NM |
| Grabowski and Prayson, 2015 2 16 | 12 | 10 / 18 / M; 2 / M; 9 / M; 3 / M; 4 / M; 30 / F; 1 / F; 17 / F; 23 / M; 23 / M | NM | 6 LC sin FAE 1 con C y FAE 2 LC con FAE 1 Desconocido |
| Roux et al., 2018 9 1 | 1 | 1 / 11 / M | Oxcarbazepina Lacosamida | Control C y FAE |
| Caso actual 1 | 1 | 1 / 15 / F | Carbamazepina Clobazan Alparazolam Clorpromazina Orcarbazepina Topiramato | LC sin FAE |
*LC: libre de crisis; NM: no se menciona FAE: fármacos antiepilépticos; C: crisis.
Los estudios neurofisiológicos realizados a pacientes con MA esporádica han demostrado que la actividad epileptogénica puede provenir de la propia lesión, de la corteza perilesional o encontrarse en un área alejada a esta. (8)(15)(28) Esto pudiera explicar la persistencia de las crisis epilépticas en el postoperatorio de pacientes a los que solo se les ha realizado lesionectomía. (13) En el caso que se presenta, la MA y la corteza cerebral que la rodeaba eran epileptogénicas.
La resección quirúrgica total de la lesión es el tratamiento de elección y curativo en la mayoría de los casos. Entre un 43-68% de los casos alcanza una mejoría significativa de las crisis posterior a la cirugía. El 70-80% de los pacientes mantiene los FAE. (18) De los 16 casos recogidos en la tabla 1, el 81.2% está libre de crisis, 7 de ellos sin FAE (43.7%).
En conclusión, la MA es una lesión benigna e infrecuente. El diagnóstico debe ser descartado en pacientes jóvenes con crisis epilépticas intratables a la medicación con masa calcificada en las imágenes de TAC y dificultades en caracterizarla en las IRM. Ante la sospecha de MA, se recomienda realizar estudios de ECoG intraoperatoria por la posible asociación con DCF en la neocorteza perilesional lo que pudiera incrementar su incidencia y el conocimiento sobre estas dos lesiones. El estudio histológico proporciona el diagnóstico definitivo.