









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las calcificaciones ganglio basales simétricas bilaterales, conocidas como síndrome de Fahr, son un hallazgo infrecuente en las neuroimágenes, se puede encontrar en aproximadamente el 1% de las tomografías craneales realizadas (1). Esta patología se caracteriza por el depósito simétrico y bilateral de calcio en núcleo estriado (núcleo caudado y putamen), tálamo lateral, cápsula interna, corteza cerebral y cerebelo (2)(3).
Descrito por primera vez en 1850 por Delacour, en un hombre de 56 años que clínicamente tenía rigidez y debilidad de las extremidades inferiores con temblor. En 1855, Bamberger, describió la entidad histopatológica de calcificaciones de los vasos cerebrales más delgados en una mujer con convulsiones y retraso mental. Posteriormente Theodore Fahr describió un caso de un paciente de 81 años con un largo historial de demencia, hipotiroidismo y evidencia de calcificaciones en la sustancia blanca, ganglios basales y arterias cerebrales pequeñas en la autopsia.
Las calcificaciones pueden ser de origen primario, genéticas o idiopáticas: Enfermedad de Fahr; o secundario (4). Dentro de las causas secundarias, se encuentran alteraciones endocrinas del metabolismo calcio/fósforo, especialmente hipoparatiroidismo y pseudo hipoparatiroidismo (5), exposición a tóxicos (plomo, monóxido de carbono), enfermedades mitocondriales, radiación intracraneal, insulto hipóxico/anóxico, infecciones, alteraciones vasculares y asociado a Síndrome de Down (1). Las calcificaciones ganglio basales idiopáticas o primarias (IBCG), se han relacionado con la mutación de los siguientes genes: SCL20A2, PDGF-B, PDGFR-B y XPR1 (6).
A pesar de ser una patología rara en la edad pediátrica es importante reconocerla, debido a que puede tener efectos deletéreos sobre la calidad y expectativa de vida.
En este reporte describimos las características clínicas e imagenológicas de un niño de 9 años con hallazgo de Enfermedad de Fahr, y su evolución terapéutica.
Caso clínico
Niño 9 años 6 meses, de sexo masculino, procedente de Venezuela, consultó a urgencias por episodio convulsivo tónico clónico generalizado de aproximadamente 4 minutos de duración, manejado inicialmente con fenitoína en institución de remisión y posteriormente se dejó mantenimiento con ácido valproico a 20 mg/kg/día. A los 3 años de edad presentó meningitis por E. coli, con posteriores secuelas en el neurodesarrollo con recuperación progresiva del habla, y la marcha; además de presencia de episodios convulsivos, por lo que recibió tratamiento durante 1 año con topiramato, sin presentar eventos convulsivos desde los 5 años de edad. Hace aproximadamente 6 meses inició con alteraciones en la marcha, temblores y distonía en la mano derecha que han sido progresivos. En cuanto al rendimiento académico había sido bueno, sin aparentes dificultades cognitivas. Presentaba alteración progresiva en la articulación del habla.
Hijo de madre primigestante, sin antecedentes de abortos, sin complicaciones durante el embarazo, controles prenatales adecuados, a término, por cesárea por desproporción cefalopélvica, peso de 2600 g sin requerimiento de reanimación, oxígeno ni hospitalización. No hay antecedentes de consanguinidad en padres, ni abuelos. Sin antecedentes familiares genéticos o metabólicos conocidos.
Al ingreso el niño se encontraba alerta, orientado, colaborador. Sin alteraciones de sensibilidad, ni de fuerza, excepto en la mano derecha donde se evidencia postura distónica, con marcado temblor de intención y limitación funcional; retracciones isquiotibiales bilaterales y marcha agazapada. Antropometría normal para su edad con una talla de 140 cm., en 0.56 desviaciones estándar para edad y peso de 36 kg con 0.89 desviaciones estándar y un IMC de 0.84. No se encontraron alteraciones al examen oftalmológico. Fonoaudiología encuentra alteración en el uso de conectores en el habla y dificultades en la articulación por debilidad del orbicular de los labios. Se realizó valoración del perfil cognitivo y del desarrollo de las funciones corticales emergentes por un profesional en neuropsicología. Dado el compromiso motor del paciente, en la aplicación de la Escala de Inteligencia de Wechsler para niños (WISC-IV) (Tabla 1) (7) y en los subdominios de atención, memoria, lenguaje y habilidades escolares de la Batería de Evaluación Neuropsicológica Infantil (ENI) (Tabla 2) (8), se prescindió de las pruebas manipulativas. Debido a dicha limitación, en el WISC - IV, solo se contemplaron las escalas de Comprensión verbal y Memoria de trabajo, en las cuales obtuvo un índice compuesto de 71 y 77 respectivamente, ubicándose en el rango inferior - limítrofe (Tabla 1). En el caso de la ENI, se observa que la atención auditiva se halla medianamente preservada. En la memoria a corto plazo, hay labilidad en la codificación y evocación diferida para información audioverbal. En el lenguaje, a pesar de presentar un trastorno del habla, logra denominar, designar y seguir instrucciones. Su ejecución en lectura también es funcional. En conclusión, los datos indican un perfil cognitivo y de las funciones corticales emergentes que se sitúa entre un desempeño limítrofe y el déficit cognitivo leve.
Tabla 1 Resultados del paciente con la Escala de Inteligencia Wechsler para niños WISC-IV.
| SUBPRUEBA | PUNTUACIONES ESCALARES | |||
| Diseño con Cubos | NA | |||
| Semejanzas | 6 | |||
| Retención de dígitos | 6 | |||
| Conceptos con dibujos | 8 | |||
| Claves | NA | |||
| Vocabulario | 4 | |||
| Sucesión de números y letras | 6 | |||
| Matrices | 6 | |||
| Comprensión | 5 | |||
| Búsqueda de símbolos | NA | |||
| CV | RP | MT | VP | |
| Suma puntuaciones Escalares | 15 | NA | 12 | NA |
| ESCALA | SUMA P. ESCALARES | INDICE COMPUESTO | CLASIFICACIÓN | |
| Comprensión Verbal (CV) | 15 | 71 | Inferior - limítrofe | |
| Razonamiento Perceptual (RP) | NA | NA | NA | |
| Memoria de Trabajo (MT) | 12 | 77 | Inferior - limítrofe | |
| Velocidad de Procesamiento (VP) | NA | NA | NA | |
| Escala Total | NA | NA | NA |
Tabla 2 Resultados del paciente con la herramienta de Evaluación neuropsicológica infantil ENI - II.
| FUNCIONES COGNOSCITIVAS ENI | ||||
| Dominios | Subdominios | Puntuación Natural | Rango Percentil | Clasificación |
| Atención auditiva | Dígitos en progresión | 4 | 37 | Promedio |
| Dígitos en regresión | 2 | 5 | Limite | |
| Memoria Codificación | Lista de palabras | 24 | 9 | Limite |
| Memoria Evocación | R.E. Lista de palabras | 6 | 9 | Limite |
| R.C. Lista de palabras | 7 | 26 | Promedio | |
| R.VR Lista de palabras | 20 | 9 | Limite | |
| Lenguaje | Denominación de imágenes | 11 | 50 | Promedio |
| Designación de imágenes | 15 | 50 | Promedio | |
| Seguimiento de instrucciones | 8 | 16 | Promedio bajo | |
| Comprensión del discurso | 4 | 37 | Promedio | |
| Lectura - precisión | Palabras | 10 | 50 | Promedio |
| Oraciones | 9 | 75 | Promedio | |
| Lectura - comprensión | Oraciones | 7 | 26 | Promedio |
| Lectura en voz alta | 2 | 9 | Limite | |
| Lectura en voz baja | 2 | 26 | Promedio |
Debido a evento convulsivo sin estudios recientes se solicitó tomografía axial computarizada cerebral simple (TAC), que mostró múltiples imágenes de densidad cálcica que se ubican en áreas subcorticales en ambos hemisferios, los cuerpos estriados, los globos pálidos y en menor medida los núcleos cerebelosos (Figura 1). Con dicho hallazgo se consideró posibilidad de síndrome de Fahr, por lo que se solicitaron calcio 9.52 mg/dL, fósforo 5.4 mg/dL y PTH 42.8 pg/mL, calcio en orina espontánea menor a 1 mg/dl, TSH 4.53 uUI/ml, T4 libre: 1.50 ng/dl, fosfatasa alcalina 232 U/L, lactato 2.4 mmol/L (Referencia: 1-2.4mmol/L), todos en rangos de normalidad; función hepática preservada con AST 46 U/L ALT 20 U/L, y función renal adecuada con creatinina 0.37 mg/dL y nitrógeno ureico de 10.2 mg/dL. Dada la no sospecha de miopatía, por asimetría de la presentación clínica y no elevación de AST, no se realizó estudio de enzimas musculares. Hemograma sin evidencia de anemia, leucocitosis, ni alteraciones plaquetarias. Electrocardiograma sin alteraciones, ecocardiograma normal. Valorado también por odontopediatría, quienes encuentran hipoplasia del esmalte de forma generalizada, compatible con amelogénesis imperfecta. Debido a dificultades en el acceso por no aseguramiento en salud al ser migrante, no fue posible la realización del estudio de aminoácidos plasmáticos, ni perfil de acilcarnitinas.
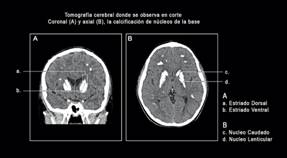
Figura 1 Tomografía computarizada de cerebro no contrastada del paciente. Se observan calcificaciones simétricas gangliobasales en imagen coronal (A) y axial (B).
Por la distonía en mano derecha, se inició Levodopa-carbidopa 1 mg/kg/día con ascenso progresivo hasta 10 mg/kg/día, sin evidencia de efectos adversos, con lo que presentó mejoría de los temblores y progresiva mejoría de la debilidad y de la distonía. Se inició programa de rehabilitación con 3 sesiones por semana de terapia física, ocupacional y fonoaudiológica.
Comentario
Reportamos el caso de un paciente pediátrico de 9 años con diagnóstico clínico de enfermedad de Fahr (primaria). Se considera primaria, habiéndose descartado alteraciones paratiroideas (la etiología secundaria más frecuente), tiroideas, infecciones, intoxicación por plomo o monóxido de carbono, sin antecedentes hipóxico-isquémicos, patologías autoinmunes, ni hallazgos sindromáticos al examen físico, tampoco compromiso multisistémico (cardiológico, renal, hepático), ni pondoestatural que pudieran sugerir patología metabólica o mitocondrial. Sin antecedentes familiares de patologías neurodegenerativas. El diagnóstico genético no se realizó.
La enfermedad de Fahr es un trastorno genético raro neurodegenerativo, autosómico dominante o recesivo, aunque también se puede presentar de manera esporádica. En el caso del paciente no se encontraron antecedentes familiares relevantes en tres generaciones. Nicolas y cols, realizaron una serie de casos de pacientes encontrando que las mutaciones podían ser familiares o casos esporádicos (6).
Las calcificaciones ganglio basales idiopáticas o primarias (IBCG), se han relacionado con la mutación de los siguientes genes: SCL20A2 (9), PDGF-B, PDGFR-B (6)(10) y XPR1 (11). La mutación del gen SLC20A2(transportador de fosfato), representa aproximadamente el 40% de los pacientes con enfermedad de Fahr. (9)(12) La mutación de la subunidad del factor de crecimiento derivado de plaquetas (PDGFB) y el receptor del factor de crecimiento derivado de plaquetas subunidad B (PDGFRB) (6)(10)(12) representan el 11% y el 2% de los pacientes con enfermedad de Fahr, respectivamente. Las mutaciones en el receptor de retrovirus xenotrópico y politrópico (XPR1) (11) se encontraron en aproximadamente el 2% de los pacientes. Sin embargo, dichos genes sólo explican aproximadamente la mitad de los casos de IBCG, lo que confirma la heterogeneidad genética de la enfermedad (6). En los pacientes con alguna de las mutaciones genéticas, la penetrancia es casi completa para depósitos de calcio, sin embargo, la penetrancia clínica es incompleta y puede estar alrededor del 70%. (12)
El mecanismo fisiopatológico reside en el depósito de calcio y otros minerales en el globo pálido, putamen, corona radiada, tálamo, núcleos dentados cerebelosos y sustancia blanca hemisférica y cerebelosa, que se produce primariamente en las paredes de arteriolas y capilares, posteriormente el espacio perivascular y lentamente se extiende hasta que compromete completamente la neurona (13)(14)(15)(16). Se plantea que los depósitos se deben a un metabolismo anormal de calcio, en el cerebro debido a una alteración de la barrera hematoencefálica. Las alteraciones en el transporte de hierro y la producción de radicales libres ocasionan lesión tisular, que inicia calcificaciones alrededor de un nido de mucopolisacáridos y sustancias similares. La calcificación progresiva cerca de los vasos a su vez disminuye el flujo sanguíneo y por lo tanto el ciclo vicioso de disminución de la perfusión, injuria tisular y deposición cálcica. También se ha evidenciado que puede existir un proceso repetitivo de depósito de lípidos y zonas de desmielinización (17). Se han reportado adicionalmente, niveles elevados de cobre, zinc, magnesio, hierro y alteraciones en el metabolismo de ganglios basales. En otros casos se han encontrado niveles elevados de péptido específico de sistema nervioso central, homocarnosina y niveles bajos de histidina (16).
A nivel histopatológico se pueden observar depósitos granulares y nódulos sólidos en el área comprometida y atrofia cortical leve. A nivel microscópico, se evidencian depósitos de calcio a lo largo de las paredes de arterias de pequeño y mediano calibre, con menor compromiso de las venas. Algunas veces, se pueden evidenciar calcificaciones en gota; en la zona circundante se observan cambios isquémicos y gliosis difusa. En la microscopía electrónica, el calcio aparenta material cristalino o amorfo rodeado por una membrana basal. En el citoplasma de células gliales y neuronas, se pueden ver gránulos de calcio (15)(16)
Nuestro paciente consultó por evento convulsivo, con antecedente de epilepsia secundaria a neuro infección, que se encontraba sin tratamiento anticonvulsivante; y de forma incidental en la TAC se observaron las calcificaciones ganglio basales. Morgante y cols, reportaron 3 casos de síndrome de Fahr con trastorno del metabolismo del calcio que habían tenido meningoencefalitis durante la infancia (18). Entre las estrategias neurorradiológicas, el TAC cerebral es más sensible para la detección de depósitos de calcio, que la resonancia cerebral (RM) simple, sin embargo, la RM con imagen ponderada por susceptibilidad (SWI) puede ser más sensible para los cambios tempranos (19)(20)(21) Otra herramienta útil es la tomografía computada por emisión de fotón único (SPECT) con 99m tehexametilpropilenamina oxima (99mTc-HMPAO) (22) que muestra una disminución marcada de la perfusión ganglio basal bilateral y en la corteza cerebral (23). La gammagrafía cerebral puede detectar alteraciones funcionales o metabólicas y evaluar la integridad de las vías nigroestriatal y estriado cortical. Existen otros métodos controversiales por resultados contradictorios: tomografía por emisión de positrones (PET/CT), y tomografía por emisión de fotón único con transportador de dopamina (DaT-SPECT) (18). Se ha evidenciado en algunos pacientes la disminución de la captación de glucosa en putamen, globo pálido y corteza temporal y parietal, en PET/CT con 18F-fluorodeoxiglucosa, planteando que dicha disminución en la captación podría ser secundario a la alteración de los circuitos funcionales que involucran los ganglios basales (21).
Los síntomas más frecuentes descritos son trastornos del movimiento y síntomas neuropsiquiátricos. Nuestro paciente presentaba alteraciones motoras: distonía de la mano derecha y trastorno progresivo de la marcha. Dentro de los síntomas más frecuentes en la literatura, el 56% de los pacientes presentaron trastornos del movimiento, parkinsonismo 57%, corea 19%, temblor 8% distonía 8%, atetosis 5% y disquinesia orofacial 3%; también pueden encontrarse torpeza, fatiga, marcha inestable, movimiento involuntario o calambres musculares (24)
El paciente también presentaba alteraciones progresivas del habla, hallazgo que también ha sido descrito en la literatura con habla lenta o arrastrada, disfagia, sin claridad de la prevalencia (25). La amelogénesis imperfecta detectada en el paciente se relaciona con muchos trastornos genéticos asociados con patología neurológica, sin embargo, no se encuentra literatura de la relación de amelogénesis con enfermedad de Fahr.
Nuestro paciente no presentó alteraciones psiquiátricas. Sin embargo, el perfil cognitivo y de las funciones corticales emergentes se ubicó entre un desempeño limítrofe y el déficit cognitivo leve. Esta oscilación en el rango es sugerente de la convergencia de una noxa de rápida emergencia, versus la reserva cognitiva precedida por un desarrollo neurológico relativamente normal. Se ha documentado, a partir del análisis de diferentes entidades fisiopatológicas, que una retracción anatomofuncional de los circuitos cortico-estriado-tálamo-corticales, vinculados con la actividad cognitiva, afecta procesos como la atención, la velocidad de procesamiento, la memoria, el lenguaje, entre otros (26)(27). En la serie de casos de Nicolas y cols. el 58% tenían deterioro cognitivo y el 57% síntomas psiquiátricos: incluyendo cambio de comportamiento, personalidad, psicosis y demencia, pero también se han descrito síntomas más leves como dificultad de concentración y memoria (8). En la literatura son escasas las descripciones detalladas del perfil cognitivo, lo cual constituye una fortaleza del presente reporte.
Las convulsiones se presentan de manera frecuente, asociada a la enfermedad, según lo reportado en la literatura (28). Jiménez-Ruiz reportó una serie de casos de 7 pacientes, de los cuales 4 presentaban convulsiones de diferente semiología (1). Otros síntomas menos frecuentes son cefalea crónica, vértigo, incontinencia o urgencia urinaria (25).
La variabilidad en los síntomas de presentación puede ser explicado parcialmente por la división de los ganglios basales en sistema dorsal, que juega un rol importante en funciones motoras y cognitivas; y la región ventral que participa en las funciones motivacionales. (12)
La mayoría de los casos reportados son en adultos, puesto que la edad de presentación más frecuente es entre la 4ta y 5ta década de la vida (8), con muy poca disponibilidad de literatura pediátrica. Entre los reportes de pacientes jóvenes encontramos el reporte de una paciente de 15 años, con síndrome de Fahr secundario a hipoparatiroidismo, quien debutó con presencia de convulsiones desde los 16 meses de vida (29).
El diagnóstico clínico se basa en la presencia de los siguientes criterios, que cumplía el paciente que reportamos: evidencia de calcificación bilateral de los ganglios basales en neuroimagen; presencia de disfunción neurológica progresiva; y ausencia de causa metabólica, infecciosa, tóxica o traumática.
No hay cura, ni tratamiento estandarizado para el síndrome de Fahr. El tratamiento de los pacientes está encaminado al manejo de los síntomas clínicos.
En este paciente se indicó tratamiento con carbidopa levodopa, sin evidencia de efectos adversos, y con buena respuesta clínica, consistente en mejoría de los temblores y de la distonía. A pesar de que en la literatura reportan que la respuesta a dicho medicamento es muy pobre; de forma ocasional se utilizan antipsicóticos; sin embargo, son mal tolerados por sus síntomas colaterales de extrapiramidalismo. La opción terapéutica que se está explorando radica en los antipsicóticos atípicos, aunque los resultados aún no son concluyentes (17). También se ha visto que el tratamiento con bifosfonatos puede ser prometedor, dado que tiene buena tolerancia y en una serie de casos se evidenció mejoría de los síntomas en algunos pacientes (12)
Este caso clínico, invita a lo médico a tener en cuenta esta entidad patológica como diagnóstico diferencial de pacientes con calcificaciones ganglio basales asociadas a síntomas neurológicos progresivos en cualquier espectro, e incluso en pacientes pediátricos, a pesar de lo infrecuente a esta edad. Se debe realizar un estudio completo, descartando causas metabólicas y evaluar antecedentes familiares. Aún no hay claridad sobre la fisiopatología y la correlación imagenológica con la clínica, por lo que hay amplias oportunidades de investigación sobre el tema.