









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
Las enfermedades que producen demencia son un problema global de salud pública. Se ha estimado que el coste global de la atención a los pacientes con enfermedad de Alzheimer (EA) supone más de 500 billones de dólares en estos momentos (1). Este coste irá aumentando paulatinamente a lo largo de las próximas décadas, con una estimación cercana a los 2 trillones de dólares en el año 2050. La EA es la principal causa de demencia en el mundo y está caracterizada por un cuadro progresivo de alteración cognitiva y funcional que lleva a una incapacidad severa en pocos años (2). Actualmente, no disponemos de terapias que modifiquen el curso natural de la enfermedad. Las terapias actualmente aprobadas, inhibidores de acetilcolinesterasa y memantina, tienen un efecto marginal sobre la historia natural de la enfermedad (3)(4). Bajo este escenario, la investigación en esta patología y la búsqueda de nuevas terapias es una prioridad en todo el mundo.
Desde su descripción por Alois Alzheimer (5) en 1907 dos características histológicas han servido de marcador de la enfermedad, las placas amiloides (PA) y los ovillos neurofibrilares (ON). Las PA están formadas por péptidos ß-plegados derivados de una proteína transmembrana denominada proteína precursora de amiloide (APP en inglés). A estos péptidos se les conoce como ß-Amiloide (ß-A). Hay varias formas peptídicas, siendo las más abundantes las de 40 y 42 aminoácidos. Los ON se forman por la acumulación de una proteína asociada a los microtúbulos, la proteína Tau, fosforilada de forma anómala (6). Se conoce como hipótesis de la cascada amiloide a la acumulación paulatina y patológica de del péptido ß-A en el espacio extracelular (7). Esta acumulación paulatina conformaría las PA extracelulares. Tras ello, y bajo inducción de las propias placas, o de las fibrillas de ß-A, se producirían el depósito de ON. Como hemos mencionado previamente, los ON están compuestos por proteína Tau hiperfosforilada. La proteína Tau es un componente fundamental de los microtúbulos neuronales y del citoesqueleto. Esta alteración del citoesqueleto generaría una pérdida de función neuronal y finalmente la muerte celular. Tradicionalmente, la cascada amiloide se ha apoyado en el descubrimiento de las mutaciones genéticas asociadas a la EA familiar. La mutación en el gen codificante de la APP y de las enzimas que participan en su metabolismo (codificadas en los genes presenilina 1 y 2) producen una EA de inicio precoz y con características fenotípicas e histológicas similares a la EA de inicio tardío (8). Sin embargo, en los últimos 10 años la hipótesis de la cascada amiloide ha sufrido una profunda crisis. Esto se ha debido, en gran medida, al repetido fracaso de diversas estrategias para mejorar la cognición de los pacientes mediante la reducción de la acumulación de amiloide cerebral (9).
PAPEL DEL AMILOIDE Y SU SEMEJANZA CON OTROS PEPTIDOS ANTICROBIANOS
Sorprende la escasa información que disponemos sobre el papel fisiológico del amiloide y de sus productos derivados, especialmente del ß-A, en comparación con otras proteínas como la Tau. El ß-A es generado por el procesado catabólico de la APP, fundamentalmente bajo efecto de la alfa, beta y gamma-secretasas. La ruta metabólica que implica la beta y, posteriormente, gamma-secretasa son las causantes de la producción del ß-A extracelular, especialmente en sus formas de 40 y 42 aminoácidos (10). Durante mucho tiempo se ha considerado que esta vía metabólica era, hasta cierto punto, patológica. Esta ruta permitía la acumulación extracelular de amiloide (Figura 1). Sin embargo, se estima que el ß-A está preservado evolutivamente desde hace más de 400 millones de años, apareciendo ya en los primeros peces arcaicos. Por esta y otras razones, actualmente se piensa que esta proteína debe tener una función fisiológica clara. Y el papel que parece tener más evidencias para ejercer es el de proteína asociada a la inmunidad innata (11).
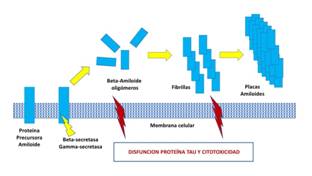
Figura 1 Hipótesis fisiopatológica propuesta para la acumulación del beta-amiloide extracelular conocida como “Cascada Amiloide”. La proteína precursora de amiloide es catabolizada por dos secretas, la gamma y beta secretasa generando péptidos beta-plegados. Estos oligómeros iniciales forman fibrillas mediante su oligomerización y finalmente placas extracelulares conocidas como placas amiloides. Se postula que tanto las placas como, especialmente, los oligómeros pueden generar citoxicidad y disfunción de la proteína Tau.
Hay diversos trabajos que establecen la actividad antimicrobiana del ß-A, inhibiendo el crecimiento tanto de bacterias gram-negativas como gram-positivas, y cultivos de hongos como Candida Albicans (12) . Así mismo, también hay reportada un efecto inhibidor sobre la infectividad del virus de la gripe A (13) y del virus herpes simple tipo 1 (14). Este comportamiento es semejante a diversos péptidos antimicrobianos (PAM) involucrados en la inmunidad innata. Los PAMs son un elemento clave en la defensa inmunológica de las células eucariotas(15). Estos péptidos pueden tener diferente tamaño, las de mayor tamaño suelen ejercer su papel mediante un rol de enzimas líticas, proteínas de unión a nutrientes clave o se dirigen frente a macromoléculas microbianas específicas. Los PAMs menores de 100 aminoácidos actúan alterando la estructura o función de las membranas celulares microbianas. Se han encontrado cientos de péptidos antimicrobianos en epitelios, células fagocíticas y fluidos corporales de animales multicelulares, desde moluscos hasta humanos. Algunos PAMs se producen constitutivamente, otros son inducidos en respuesta a infección o inflamación. Los PAMs son capaces de inhibir el crecimiento y la infectividad de bacterias, micobacterias, hongos, protozoos, virus, e incluso en algunos casos células cancerosas. En el ser humano, estos péptidos son generados mediante el proceso catabólico de precursores proteicos más grandes de forma semejante al catabolismo de la APP y la producción posterior de ß-A (16). Otra de las cualidades fundamentales de la acción de los PAMs es su tendencia a la oligomerización y generación de fibrillas. Este proceso de generación de fibrillas juega un papel fundamental en la función aglutinante de microrganismos patógenos. De esta forma, las bacterias quedan atrapadas en dicha estructura y se aglutinan perdiendo su infectividad (17). Por otro lado, también se ha descrito el efecto tóxico directo sobre las membranas celulares microbianas por los oligómeros generados en todo el proceso (18). Finalmente, los PAMs también pueden actuar estimulando y atrayendo a las células del sistema inmunológico hasta el sitio de la infección (Figura 2). Todos estos mecanismos de acción evocan claramente el proceso de generación de ß-A desde la APP, la formación inicial de oligómeros y su posterior proceso de fibrilación hasta llegar a formar PA. Evidencias sobre el efecto aglutinante del ß-A y su papel inductor de radicales libres han sido descritas recientemente (20).
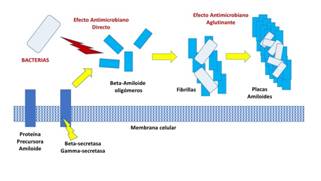
Figura 2 Hipótesis fisiopatológica propuesta al tomar el beta-amiloide como un péptido antimicrobiano. En este escenario el catabolismo de la proteína precursora de amiloide es completamente fisiológico y permite la producción de beta-amiloide en respuesta a un patógeno. Tanto los oligómeros de beta-amiloide como las placas de amiloide tendrían una función defensiva frente a bacterias o endotoxinas. En todo caso, este mecanismo inflamatorio crónico podría desencadenar neurocitoxicidad a largo plazo, bien directamente o bien por fracaso de su acción defensiva con el consiguiente daño celular producido por patógenos y/o sus productos.
Un hecho a destacar es el reconocimiento de otras amiloidosis sistémicas producidas por la disregulación de distintos PAMs en el ser humano. Hasta este momento se tiene evidencia de al menos tres amiloidopatías humanas generadas por PAMs (20). Estos cuadros son la amiloidosis corneal producida por lactoferrina (21), la vesícula seminal senil amiloide producida por el péptido semenogelina (22), y la amiloidosis atrial aislada producida por el péptido atrial natriurético (23). Los dos primeros son cuadros relativamente frecuentes y que tienen como característica habitual aparecer en sujetos añosos.
En definitiva, son crecientes las evidencias que apoyan un papel del ß-A en la inmunidad innata actuando como PAM. El papel fisiológico de estos péptidos recuerda estrechamente la ruta de producción del ß-A. Además, disponemos de evidencias directas sobre el efecto antimicrobiano directo y aglutinante del ß-A frente a microrganismos diversos. Todo ello hace conferir a la APP y sus productos, como el ß-A, un papel clave en las respuestas innatas inmunológicas del sistema nervioso central.
PAPEL DE LA NEUROINFLAMACION EN LA ENFERMEDAD DE ALZHEIMER
Tradicionalmente se ha asumido que las evidencias encontradas sobre la neuroinflamación en la EA eran elementos asociados a los estadios más tardíos de la enfermedad y como consecuencia de un epifenómeno secundario a la neurodegeneración previa. Sin embargo, actualmente existe una creciente evidencia que relaciona dicha activación del sistema inmune como un elemento clave en la patogénesis de la enfermedad (24)(25). Ya en estadios precoces de la enfermedad, como el deterioro cognitivo leve, se ha demostrado cambios inflamatorios sistémicos y en el líquido cefalorraquídeo que avalan dicha hipótesis (26)(27). El hecho más relevante es la asociación entre la EA y diversas variantes genéticas heterocigotas de los genes codificantes de los receptores expresados en las células mieloides de tipo 2 (TREM 2)(28)(29)(30)(31). TREM2 es un receptor del sistema inmunológico innato expresado en la superficie de distintas células mieloides, especialmente células dendríticas, osteoclastos y microglia. Previamente se habían descrito mutaciones homocigotas con pérdida de función asociadas a un cuadro infrecuente denominado enfermedad de Nasu-Hakola (Osteodisplasia lipomembranosa poliquística con leucoencefalopatía esclerosante). Este cuadro autosómico recesivo se caracteriza por una demencia progresiva de inicio precoz junto a quistes óseos(32).
De esta forma, los mecanismos inflamatorios propuestos en la neurodegeneración estarían liderados por una respuesta inmune innata con un papel preponderante de la microglia (24). En esta respuesta innata, a diferencia de la adaptativa liderada por linfocitos, los mecanismos inflamatorios serían responsabilidad de la microglía y de los macrófagos perivasculares. La microglía presenta tanto in vivo como in vitro reactividad frente al ß-A, especialmente en sus formas oligoméricas (33). La generación de un circulo vicioso entre la acumulación del ß-A y la activación crónica de la microglia se encuentran en muestras cerebrales de pacientes con EA (34). De esta forma, el mantenimiento de los mecanismos inflamatorios a largo plazo podría desencadenar la muerte neuronal como elemento final fisiopatológico.
MICROBIOTA, BACTERIAS Y ENFERMEDAD DE ALZHEIMER
El papel de la microbiota intestinal y bucal tiene un creciente interés en el estudio de las enfermedades neurológicas y psiquiátricas. A esta relación se le ha denominado eje microbiota-intestino-cerebro (35). Aunque los microganismos más comunes en el intestino son los lactobacillus y las bifidobacteria, existen miles de especies distintas en la microbiota intestinal de un individuo adulto sano. Este complejo y rico mosaico de microrganismos se ha asociado a distintos efectos sobre el sistema nervioso central. Los mecanismos patológicos involucrados son diversos. La producción de sustancias (monoaminas y aminoácidos) por las bacterias intestinales pueden pasar al sistema linfático y/o vascular afectando a la función del cerebral a distancia. Algunas de estas sustancias pueden comportarte como falsos neurotransmisores o neuromoduladores generando cambios conductuales(36). Otra hipótesis es la relación entre el nervio vago y el intestino. Sus proyecciones terminan en todo el tubo digestivo y enlazan estas estructuras con el tronco cerebral, tálamo y corteza(37). Finalmente, también cabe la posibilidad de un efecto directo de las bacterias o sus toxinas sobre el sistema nervioso central. Esta última posibilidad no se había planteado hasta épocas recientes por la presencia de la barrera hemato-encefálica y la posición de privilegio de las estructuras cerebrales a las infecciones.
La demostración de un efecto directo de las modificaciones de la microbiota en la fisiopatología de la EA está basado, sobre todo, en estudios sobre animales. Se ha demostrado una reducción relevante de la presencia de ß-A en animales criados en ambientes asépticos; y estos animales vuelven a generar ß-A cuando son expuestos a un ambiente con patógenos (38). El uso de tratamientos antibióticos en modelos animales tiene efectos dispares. Así, el uso de ampicilina en ratas conduce a trastornos conductuales y alteración de la memoria espacial. En cambio, estos efectos son revertidos mediante la administración de probióticos como lactobacillus (39). El uso de rifampicina en modelos murinos de EA reduce la acumulación de ß-A y los niveles de citoquinas inflamatorias (40). Un hallazgo similar se ha descrito con el uso de minociclina en igual modelo, comprobando además una mejoría cognitiva en la memoria espacial(41)(42). Por otro lado, también se ha descrito una reducción de la fosforilación de la proteína Tau y de la activación de la microglía con el uso de rapamicina en modelos similares(43). A pesar de estas evidencias, los resultados de los ensayos clínicos que emplearon antibióticos no han obtenido resultados satisfactorios. Así, tras un resultado inicial prometedor (44), un ensayo aleatorizado con 406 pacientes con EA leve o moderada no demostró beneficio con el uso de rifampicina junto a doxiciclina para retrasar el deterioro cognitivo en este grupo de sujetos(45).
ENDOTOXINAS Y ENFERMEDAD DE ALZHEIMER
Las endotoxinas son un tipo de liposacárido que forma parte de la membrana externa de las bacterias gram-negativas(46). Suele estar conformado por un lípido A y una cadena variable de glúcidos conocida como antígeno O. Las endotoxinas pueden ser segregadas al espacio extracelular por las bacterias gram-negativas y generar una potente reacción inmunológica tanto innata como adaptativa. Si los niveles de entoxinas son muy elevados pueden conducir a la muerte del sujeto por un shock séptico. En el caso de persistencia de niveles reducidos pero mantenidos en el tiempo, se puede generar un estado crónico inflamatorio asociado a diversas enfermedades crónicas(47).
Disponemos de diversas evidencias en modelos animales sobre el efecto de las endotoxinas en el desarrollo de neuroinflamación crónica y fenómenos neurodegenerativos(48). La administración mantenida de endotoxinas junto a ß-A en corteza frontal y parietal de simios produce una aceleración del proceso amiloidogénico(49). También hay descrito un efecto sobre la fosforilación y agregación de la proteína Tau tras la inyección intrahipocampal de endotoxinas(50). Las endotoxinas son trasportadas en el flujo plasmático por la APOE, sugiriendo que la presencia de APOE4 podría hacer más sensible al sujeto a su efecto patogénico o a la eliminación de las mismas(51)(52). Finalmente, se ha demostrado la presencia de endotoxinas en las PA en muestras de tejido cerebral de pacientes con EA(53). Un estudio encontró niveles plasmáticos de endotoxinas hasta tres veces superiores entre los pacientes con EA frente a los controles sanos(54).
PERIODONTITIS Y ENFERMEDAD DE ALZHEIMER
Desde hace tiempo es conocida la relación epidemiológica entre la enfermedad periodontal crónica y la presencia de trastornos cognitivos en edades avanzadas(55)(56). Se ha descrito una relación entre esta patología y la acumulación de ß-A en cerebros de sujetos cognitivamente sanos, empleando como herramienta el PET con marcador para amiloide(57). Además, un estudio observacional entre pacientes con EA con periodontitis crónica mostró un curso más deletéreo comparado con los pacientes que no presentaban periodontitis durante los 6 meses de observación(58).
Recientemente, la publicación del grupo de Dominy y cols(59) sobre el papel de la Porphyromonas gingivalis (PG) en la fisiopatología de la EA ha revolucionado este campo de estudio. La infección crónica bucal por PG es uno de los principales patógenos de la periodontitis crónica. Es conocida la bacteriemia transitoria por PG en ciertas circunstancias como el cepillado de dientes o la masticación, y especialmente durante la realización de procedimientos odontológicos(60). Un estudio reciente, empleando sondas genómicas en la detección de bacterias en las placas arterioscleróticas, documentó la presencia de genoma de PG en prácticamente todos los pacientes estudiados(61). La PG es una bacteria gram-negativa anaerobia que tiene como característica patogénica la secreción al espacio extracelular bacteriano de una proteasa de gran virulencia conocida como gingipaína. Las gingipaínas son un conjunto de cistein-proteasas que tienen un papel crítico en la colonización e inhibición de las defensas del huésped. En el estudio referido previamente de Dominy y cols(59) se analizan en diversos experimentos el papel de la PG y de las gingipaínas en el desarrollo de la fisiopatología de la EA. En un análisis de muestras de corteza temporal medial se demostró una diferencia significativa en la detección de gingipaínas entre 29 sujetos con EA (96-91%) frente a 29 controles sanos (52-39%). La detección de genoma de PG fue del 100% en 3 muestras corticales de pacientes con EA frente al 83,3% en 6 muestras de controles sanos. También se realizó un análisis de detección de genoma de PG en el líquido cefalorraquídeo de 10 pacientes con EA demostrando positividad en 7/10; en el mismo grupo el análisis en saliva fue positivo en 10/10. En el mismo trabajo se analizaron diversos experimentos sobre el papel de las gingipaínas en el metabolismo de la de ß-A y de la proteína Tau. Así, se demostraba la presencia de gingipaínas en los ON y un efecto in vitro de fragmentación de la proteína Tau que podría desencadenar su fosforilación y agregación. Respecto a la ß-A, se demostraba el efecto antimicrobiano frente a la PG de dicho péptido, espacialmente de los péptidos de 42 aminoácidos. Todo este corpus de conocimiento se aplicó al desarrollo de inhibidores de la gingipaína como potenciales neuroprotectores. En el mismo trabajo, se muestran resultados de una pequeña molécula con efecto inhibidor irreversible de dichas proteasas. Estos inhibidores de gingipaína previenen la muerte celular in vitro y evitan los procesos neurodegenerativos en un modelo murino infectado por PG. En definitiva, los resultados publicados abren la posibilidad a una línea terapéutica innovadora y disruptiva en el campo de las enfermedades neurodegenerativas, especialmente en la EA. Actualmente hay en marcha un ensayo clínico fase 2/3 con una molécula inhibidora de la gingipaína, conocida como COR388, entre pacientes con EA(62).
¿LAS ENFERMEDADES NEURODEGENERATIVAS TIENEN UN ORIGEN INFLAMATORIO?
Las hipótesis previas sobre un origen causal de la EA debido a la acumulación paulatina y primaria del ß-A parecen hoy en día superadas. Las evidencias actuales centran a la APP y sus productos catabólicos, especialmente el ß-A, como un elemento de la respuesta inmune innata. La relación entre neuroinflamación y neurodegeneración son complejas y establecen un escenario de sinergias entre los distintos actores que participan en estos fenómenos. El nuevo modelo teórico (Figura 3) establece como causa inicial la alteración de la microbiota a nivel intestinal o bucal, generando una entrada de microrganismos o de sus productos en el organismo(24)(35)(46). Las endotoxinas de las bacterias gram-negativas son liposacáridos con un potente efecto inmunógeno tanto para el sistema inmunológico innato como adaptativo. Estas endotoxinas tienen efecto amiloidogénico y generan disfunción de la proteína Tau en modelos animales(47)(48). Uno de los hechos más controvertidos de la casacada amiloide ha sido la presentación del acúmulo de ß-A desde décadas previas al inicio de los síntomas, así como su mala correlación con la sintomatología del paciente. La confirmación del papel del ß-A como una proteína clave en la defensa innata inmunológica sería una evidencia sólida de la hipótesis inflamatoria de las enfermedades neurodegenerativas. En ese escenario, la amiloidogénesis estaría directamente relacionada con la defensa frente al efecto de las endotoxinas bacterianas o de las propias bacterias. Esta acumulación de ß-A tendría un papel protector en las primeras fases de la enfermedad; sin embargo, finalmente la persistencia de los fenómenos inflamatorios podría generar neurotoxicidad. Este efecto deletéreo sobre las neuronas podría estar ligado a la toxicidad de los oligómeros del ß-A, o bien a la toxicidad directa de las endotoxinas una vez superada la defensa innata inmunológica. Los recientes hallazgos del efecto directo de las gingipaínas de la PG (una potente endotoxina bacteriana) sobre la proteína Tau, establece una conexión directa entre ambos fenómenos(59).
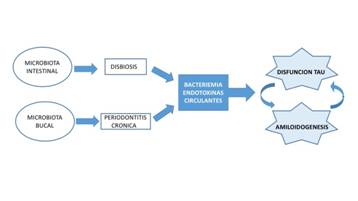
Figura 3 Hipótesis fisiopatológica neuroinflamatoria de la enfermedad de Alzheimer. La alteración del equilibrio en la microbiota intestinal o bucal generaría un paso al torrente sanguíneo y tejidos de bacterias locales. Hay evidencias que apoyan un efecto amiloidogénico de ciertas endotoxinas bacterianas de gram-negativas. Este efecto ha sido estudiado recientemente con las gingipaínas, una endotoxina producida por una bacteria anaerobia asociada a la periodontitis crónica denominada Porphyromonas gingivalis. Las gingipaínas parecen inducir la amiloidogenesis y bajo ciertas condiciones disfunción de la proteína Tau(59).
CONCLUSIONES
La EA es la principal causa de demencia en todo el mundo y su causa es desconocida. Aunque las dos principales lesiones observables en los cerebros de los pacientes, las PA y los ON, fueron descritas hace más de 100 años, aún no se conoce su significado fisiopatológico en su totalidad. En los últimos años, existen crecientes evidencias que apoyan un origen neuroinflamatorio para las enfermedades neurodegenerativas y la EA en particular. Este fenómeno inflamatorio estaría liderado por los mecanismos derivados de la inmunidad innata y restringido en gran parte a algunas regiones del sistema nervioso central. El papel del ß-A como un sistema de defensa innata frente a distintos patógenos bacterianos es creciente y podría revelarse como un mediador inflamatorio no causal de la enfermedad. Finalmente, la demostración de un efecto directo de la PG en el desarrollo de neurotoxicidad mediada por uno de sus productos, las gingipaínas, supone una línea de investigación muy esperanzadora en cuanto al desarrollo de un tratamiento modificador de la enfermedad. El tiempo será quien nos dé o nos quite la razón.