









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkAntecedentes
El complejo esclerosis tuberosa (CET) es una enfermedad neurocutánea de presentación variable, transmitida por herencia autosómica dominante y con prevalencia entre 1/6,000 a 1/12,000 nacidos vivos.(1)(2)(3) Sus manifestaciones más comunes son lesiones de varios tipos en piel, cerebro, riñones, pulmones y corazón, con tumoraciones que pueden resultar en disfunción de órganos al sustituir el parénquima normal.(1)
Aproximadamente dos tercios de los casos se deben a mutaciones espontáneas en los genes TSC1 (hamartina, cromosoma 9q34.13) y TSC2 (tuberina, cromosoma 16p13.3), que poseen un efecto de supresión tumoral.(1)(2)(3) Sin embargo, 15 a 25% de los casos no poseen mutaciones en dichos genes.(3)
El cuadro clínico se manifiesta clásicamente por la Triada de Vogt (retraso mental, epilepsia y adenoma sebáceo),(3) siendo la epilepsia la manifestación más común con espasmos infantiles de inicio en los primeros meses de vida.(4)(5) No obstante, la mitad de los pacientes presentan inteligencia normal y un cuarto de ellos no desarrolla epilepsia.(4)(5) El diagnóstico definitivo se basa en el hallazgo de manifestaciones clínicas mayores y menores,(1)(2)(3) considerando además hallazgos imagenológicos y de pruebas genéticas (Tabla 1).(1)(2)(3)(6)(7) Este diagnóstico puede ser difícil para los médicos de atención primaria en las comunidades rurales.
Tabla 1 Criterios diagnósticos para CET (versión 2012)3 y comparación con el caso presentado.
| CRITERIOS MAYORES | SI | NO |
|---|---|---|
| Maculas hipomelanóticas (≥3, al menos 5mm de diámetro) | ||
| Angiofibromas (≥3) | ||
| Fibromas ungueales (≥2) | ||
| Parches de Shagreen | ||
| Hamartomas retinales | ||
| Displasias corticales (incluye tubérs y líneas radiales de migración en sustancia blanca) | ||
| Nódulos subependimarios | ||
| Astrocitomas subependimales de células gigantes | ||
| Rabdomiomas cardíacos | ||
| Linfangio-leiomatosis* | ||
| Angiolipomas (≥2)± | ||
| CRITERIOS MENORES | SI | NO |
| Lesiones “confeti” en la piel | ||
| Lesiones en esmalte dental (>3) | ||
| Fibromas intraorales (≥2) | ||
| Parches acrómicos en retina | ||
| Múltiples quistes renales | ||
| Hamartomas no renales |
*Una combinación de linfangio-leiomatosis y angio-miolipomas sin otras características no reúne criterios para un diagnóstico definitivo.1-3 El diagnóstico definitivo de CET se hace con a) dos criterios mayores, o b) un criterio mayor con dos o más criterios menos, o c) identificación de una variante heterocigótica patogénica en los genes TSC1 o TSC2.
Reportamos un caso atípico de CET con espasmos infantiles de inicio tardío diagnosticado por una red piloto de telemedicina conectando médicos de atención primaria de una zona rural con especialistas en otra ciudad de Honduras y un hospital de Estados Unidos.
Caso Clínico
Se nos refirió paciente femenina de 22 meses de edad procedente de un municipio rural del Departamento de Olancho. La niña estaba previamente sana e inició síntomas a los 18 meses de edad, presentando episodios de oculogiros sin pérdida de conciencia, a veces seguidos de somnolencia hasta por una hora. Dos semanas después se agregaron episodios de extensión simétrica, brusca y breve de ambos miembros superiores, los cuales continuaron ocurriendo múltiples veces al día y sin alteración de conciencia (Figura 1.). La paciente había recibido tratamiento con ácido valproico y levetiracetam sin lograr control de crisis.
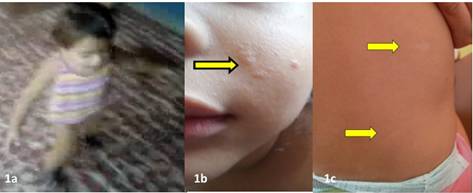
Figura. 1 a) Episodios de elevación brusca breve en extensión de ambos miembros superiores, extracto de vídeo casero a los 18 meses de edad; b) Angiofibromas en mejilla izq.; y 1c) Manchas hipomelanóticas en tórax posterior.
El caso fue conocido inicialmente para una tele-interpretación de encefalograma digital hecha hasta la ciudad de Tegucigalpa. La niña había sido llevada a la ciudad para realizar una resonancia magnética e interconsultas, pero no se había establecido un diagnóstico. Ante las dificultades para su manejo, los médicos de atención primaria tratantes interconsultaron a través de la red piloto de telemedicina que conectaba dos estaciones en Honduras (Tegucigalpa capital de Honduras y la Clínica Buen Pastor del Municipio rural de Santa María del Real en el Departamento de Olancho) con la Universidad Médica de Carolina del Sur (MUSC) en Estados Unidos. Los aspectos técnicos de la red han sido descritos por separado.(8)
Todos los exámenes fueron digitalizados para la teleconsulta, en la cual participaron especialistas en pediatría, neurología y genética de MUSC. Se utilizó cámaras de examen y estetoscopio digitales, así como cámara con luz ultravioleta para revisión de las manifestaciones dermatológicas de la paciente. También se disponía de videos caseros de crisis epilépticas grabadas con celular por la madre, quien firmó el consentimiento informado respectivo.
Además de los episodios descritos, se refirió desde el nacimiento notaron la presencia de pápulas en la cara de la paciente, así como lesiones hipomelanóticas pequeñas en el tórax posterior. Estas aumentaron en tamaño y número con la edad. No se reportó anormalidades del crecimiento y desarrollo, ni antecedentes perinatales patológicos o traumáticos, pero se reportó que un tío paterno padecía de epilepsia.
Al examen físico tele-transmitido se encontró a una niña activa y colaboradora, con un peso de 16kg (percentil 90), perímetro cefálico de 47cm (percentil 25) y talla de 97cm (percentil 75). Al examen segmentario no se encontró anormalidades en cabeza, cuello, cardiopulmonar, abdomen ni extremidades. En piel se encontraron lesiones papulares, lisas, blandas, de igual color que la piel que medían de 1 a 3mm de diámetro, distribuidas asimétricamente en región nasal, perinasal y en mejilla izquierda. También se documentaron lesiones hipomelanóticas, alargadas, en forma de hoja de fresno de 3mm-12mm de diámetro, localizadas en región inferior del tórax posterior (Figura 1.).
No se observó anormalidades en lechos ungueales ni en el esmalte dental. El desempeño crecimiento y desarrollo de la paciente eran normales para su edad, sin evidencia de retraso mental. El examen neurológico remoto no mostró anormalidades, el fondo de ojo fue reportado como normal y el desempeño psicomotor durante la evaluación fue el esperado para la edad.
A pesar de las limitaciones financieras, la paciente había sido llevada a la ciudad de Tegucigalpa para realizar estudios de neuroimagen, EEG y consultas con varios médicos. El EEG interictal mostró actividad epiléptica focal y generalizada sin hipsarritmia (Figura 2.). No se reportó eventos clínicos durante este registro.
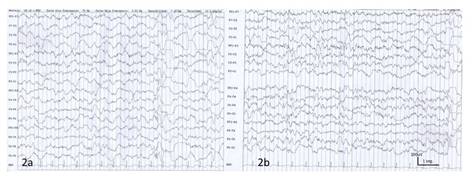
Figura 2 EEG interictal con actividad epiléptica multifocal, frontocentral bilateral y también generalizada en forma de complejos punta onda irregulares, además con lentificación focal temporal izquierda.
El reporte de tomografía cerebral tomada a los 20 meses de edad describía varias lesiones hipodensas corticales de pequeño tamaño acompañada de una calcificación temporo-medial izquierda y al menos dos lesiones nodulares adosadas a ambos atrios ventriculares. Las imágenes no estaban disponibles. La resonancia magnética (RM) de cerebro tomada a los 22 meses de edad mostró dos lesiones en la secuencia T1 sugestivas de túbers en la regiones temporal izquierda y fronto parietal derecha; así como otras hiperintensidades evidentes en la técnica FLAIR, todas ellas corticales, de pequeño tamaño, ligeramente triangulares o rectangulares siguiendo la morfología de los giros en donde se localizaban. Además se observaron nódulos subependimarios menores a 5mm adosados a las paredes de los atrios ventriculares, que reforzaron con gadolíneo (Figura 3.).
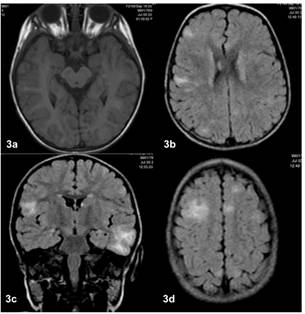
Figura 3 RM con lesiones compatibles con túbers en la región fronto parietal derecha y temporal izquierda (3a-c), así como otras hiperintensidades múltiples evidentes en la técnica FLAIR (3b-d). Además se observaron nódulos subependimarios adosados a las paredes de los atrios ventriculares (3b).
El consenso de los consultantes fue que el caso cumplía criterios diagnósticos de CET, con 4 criterios mayores: manchas hipomelanóticas, angiofibromas faciales, displasia cortical con túbers y nódulos subependimales (Tabla 1).(1)(2)(3) Sin embargo, el caso no tenía la presentación clásica de epilepsia con espasmos infantiles que inician antes de los 12 meses de edad, ni tenía retraso psicomotor (Tabla 1). Se readecuó el manejo terapéutico iniciando lamotrigina, la cual permitió pronto control de crisis casi total desde la dosis inicial. Se incorporó un plan de seguimiento a largo plazo con evaluaciones cada 6 meses para detección y manejo precoz de complicaciones.
Discusión
Este caso ilustra cómo la telemedicina puede ayudar a pacientes neurológicos con diagnósticos complejos y que viven en zonas rurales. La paciente tenía una presentación poco frecuente de esclerosis tuberosa, sin retraso mental y con espasmos infantiles de inicio tardío y refractarios.
En Honduras hay poca estadística sobre el CET. Una serie sobre síndromes neurocutáneos en un hospital público y dos clínicas privadas de neurología en Tegucigalpa encontraron que el CET era el segundo entre los demás síndromes (17% de frecuencia).(9) Otro estudio en Costa Rica reportó que las principales manifestaciones de CET en población pediátrica fueron calcificaciones, tubérculos corticales, manchas hipomelanóticas y angiofibromas faciales.(10)
En el 2012, el Consenso Internacional de CET incluyó un criterio genético, con presencia de mutaciones en los genes TSC1 o TSC2.(2)(6) En países como el nuestro, donde el acceso a pruebas genéticas es limitado, la clínica, la electroencefalografía y la neuroimagen continúan siendo el pilar para la sospecha y diagnóstico de la mayoría de enfermedades neurológicas, incluyendo el CET.
La triada del CET incluye retraso mental,(1)(3) pero la paciente no presentó esta condición a la edad evaluada a pesar del número, localización y tamaño de sus lesiones cerebrales. Se conoce que las tuberosidades de gran tamaño, aunque sean poco numerosas se relacionan con sintomatología clínica más severa como crisis epilépticas, retraso mental y comportamiento autista. En cambio, las tuberosidades de pequeño-mediano tamaño, aunque sean muy numerosas, se asocian a manifestaciones menos severas.(11)(12)
Las crisis epilépticas ocurren generalmente en 72-85% de pacientes con CET y un 80% de ellos comienzan en los tres primeros años de vida.(1)(2)(3)(7) Además de los espasmos infantiles, pueden ocurrir otro tipo de crisis epilépticas como tónicas, atónicas, tónico-clónicas, crisis parciales y ausencias atípicas. Dos tercios de los casos pueden evolucionar a una epilepsia refractaria.(13)(14)
En el CET, lo típico es que los espasmos infantiles ocurran en un 30% de los casos y con un pico de inicio entre los 4 a 6 meses de edad,(13) más temprano que en la paciente evaluada. Los espasmos infantiles tienen otras causas además del CET y ocasionalmente pueden aparecer en edad tardía (después del año de edad) cuando se asocian a una encefalopatía epiléptica, que se acompañan de crisis refractarias y déficits motores y cognitivos severos.(14)
En la paciente evaluada los espasmos aparecieron tardíamente a los 18 meses como parte de una epilepsia refractaria al valproato y levetiracetam. Los episodios de oculogiros pudieron deberse a ausencias atípicas o a espasmos, pero no fue posible estudiarlos con video-EEG. Cuando los casos de CET son típicos con espasmos de inicio temprano, el patrón electroencefalográfico incluye un aspecto caótico, mezclando ondas lentas, ondas agudas, puntas y desincronización del ritmo de fondo.(1)(4)(13) A esto se denomina hipsarritmia, elemento que tampoco observamos en la paciente.
Las manifestaciones dermatológicas que hacen sospechar CET son las lesiones hipomelanóticas que se encuentran en un 90% y los angiofribromas en un 75% de los casos.(7) En nuestra paciente, las manchas hipomelanóticas y angiofibromas faciales aparecieron sutilmente desde el nacimiento, pasando desapercibidas. Se ha descrito que estas manifestaciones cutáneas pueden ser difíciles de identificar en los primeros años de vida.(1)
Como parte del seguimiento establecido para la paciente, se indicó a los médicos primarios la evaluación periódica por complicaciones y comorbilidades propias del CET. La evaluación de función renal y el USG abdominal de la paciente fueron normales, pero se deberá vigilar por angio-mio-lipomas y quistes renales, que tienen una frecuencia de 80% y 50% respectivamente.(1)(3)(4)(12) Estas anormalidades tienden a crecer en número y tamaño con la edad. Es recomendable realizar RM abdominal cada uno a tres años y pruebas de función renal anuales o según el caso.(7)
La salud cardíaca también debe vigilarse en el CET. Los rabdomiomas cardíacos ocurren en 60% de los casos con CET y pueden detectarse en un control prenatal de rutina después de las 20 semanas de gestación, aunque se han detectado casos desde las 17 semanas gestacionales. Generalmente, estos rabdomiomas son asintomáticos, pero pueden asociarse a arritmias y al síndrome Wolff- Parkinson White.(1)(3)(7)
Alrededor de la mitad de casos de CET pueden presentar linfangio-leiomiomatosis. Aunque suelen ser asintomáticos, las formas más severas pueden afectar la función pulmonar. Otras manifestaciones de CET que deben vigilarse incluyen fibromas intraorales, angio-miolipomas hepáticos, hamartomas retinianos y conglomerados acrómicos retinales.(7)
Aunque no se cuenta con un tratamiento curativo, el control temprano de las crisis epilépticas es importante. Se recomienda la vigabatrina como primera elección cuando predominan los espasmos infantiles(7), con terapia hormonal adrenocorticotrópica como segunda línea. En Honduras no contamos con estos fármacos, pero la paciente mejoró con lamotrigina llegando a controlar casi el total de las crisis.
En cuanto a las lesiones dermatológicas, se recomienda el láser, la escisión quirúrgica o las preparaciones tópicas con mTOR para los angiofibromas faciales, pues las lesiones pueden llevar a desfiguración, hemorragias y afectación psicológica.(3)(7)
La rapamicina y el everolimus son opciones de tratamiento para prevenir la severidad de la enfermedad. Su mecanismo es inhibidor de los complejos proteínicos mTOR,(15) regulando el crecimiento y proliferación celular, pudiendo en consecuencia disminuir el tamaño de los tumores como astrocitomas y angiomiolipomas renales.(15)(16) La cirugía sería opción en casos selectos. No se cuenta con estos medicamentos en Honduras y no fue posible asistir en otros tratamientos por migración de la familia a otro país.
Conclusión
El caso discutido tenía un patrón de presentación atípico lo cual ha sido descrito ocasionalmente en el CET, especialmente desde que se hace más diagnóstico molecular.(13)(17)(18) El caso también es atípico por la aparición de espasmos epilépticos de inicio tardío, lo cual es una situación infrecuente pero ya reportada en la literatura.(14)(19) La telemedicina fue una herramienta importante para la evaluación de este caso, entre las primeras experiencias exitosas de atención neuropediátrica y neurogenética realizadas por teleconsulta en Honduras.(8)(20)
La inclusión de tecnologías como la telemedicina aplicada a la atención neurológica será de gran apoyo en las zonas rurales en Honduras y otras regiones latinoamericanas.(20)(21) Arturo Lo Famulari afirmó en esta Revista en 2012 que “la telemedicina ya es una realidad entre nosotros y desplegará ante nuestros ojos sus alas multicolores al mejor estilo de un pavo real”.(22)
Como al mencionado autor, a los neurólogos, nos interesa que la urgencia por reducir los costos operativos y por brindar acceso a atención neurológica no disminuya la importancia de la historia clínica por la excesiva confianza en la tecnología. Esto será un nuevo reto para la neurología en América Latina.