









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Se estima que los trastornos de Tics afectan al 4-20% de la población general y en algunos casos se acompañan de estrés físico y social debilitante (1). El síndrome de Tourette (ST) es un trastorno neurológico y conductual de inicio en la infancia caracterizado por combinaciones de tics motores y tics vocales que persisten por más de 1 año (1). En la población general, 0.5-0.7% de las personas se ven afectadas por ST en algún momento de sus vidas, con una mayor prevalencia en hombres que en mujeres (3: 1) (2)(3). La edad media de aparición en los niños es a partir de los 6 años, y los tics alcanzan el pico de su gravedad aproximadamente entre los 10.6 ± 2.6 años de edad (4)(5).
El diagnóstico del ST no implica ninguna prueba de laboratorio. Se realiza a partir de la evaluación clínica siguiendo las pautas del Manual Diagnóstico y Estadístico de los Trastornos Mentales (Diagnostic and Statistical Manual of Mental Disorders, abreviado DSM-IV) que incluye tres componentes: 1) presencia de tics motores y vocales durante más de un año, aunque no es necesario que ocurran simultáneamente. 2) el inicio del tic debe ser antes de los 18 años. 3) los tics no deben ser causados por la ingesta de sustancias u otra afección médica. Un historial familiar de síntomas neurológicos similares respalda el diagnóstico del ST. El diagnóstico del ST se puede ver reforzado por presencia de comorbilidades que incluyen trastorno obsesivo-compulsivo (TOC) y trastorno por déficit de atención con hiperactividad (TDAH) (5). El ST se puede aislar de otros trastornos del movimiento hipercinético como corea, distonía, atetosis, mioclono y discinesia paroxística por sus dos cualidades únicas: capacidad de suprimir los tics y las sensaciones premonitorias que a menudo los preceden (6)(7).
La presentación de los tics motores puede clasificarse como simple, compleja y plus; los tics simples involucran un grupo muscular, como parpadeo (8). Los tics complejos implican movimiento de múltiples grupos musculares y pueden ser una secuencia de movimientos o una combinación de tics simples, incluyen muecas combinadas con giro de la cabeza o patadas (8); los tics plus incluyen comorbilidades del comportamiento. Los tics vocales a menudo surgen después del inicio de los tics motores y se pueden dividir en simples y complejos. Los tics vocales simples son la producción de varios sonidos y ruidos, como aclarar la garganta, toser u olfatear; mientras que los tics vocales complejos, pueden ser lingüísticamente significativos, incluidos coprofenómenos (coprolalia, coproraxia y coprografía), y otros fenómenos que contemplan la imitación (ecolalia, ecopraxia y ecografía), la repetición (palilalia, palipraxia y paligrafía), así como la existencia de conductas más amplias socialmente inaplicables (3).
Los tics se presentan de manera creciente y se exacerban en períodos de ansiedad, estrés, excitación o enojo, y disminuyen o cesan durante el sueño, el descanso o la concentración(2)(8). La edad parece ser un factor determinante de la gravedad, ya que la prevalencia de tics tiende a disminuir desde la infancia hasta la adolescencia y adultez temprana (3). La presentación clínica variable del ST puede contribuir a un retraso en el diagnóstico, o diagnóstico erróneo.
Se conoce que el ST tiene una arquitectura genética heterogénea, donde múltiples genes contribuyen al desarrollo de la enfermedad. Los estudios con gemelos han encontrado una tasa de concordancia de 53% en gemelos monocigóticos y 8% en gemelos dicigóticos; en estudios familiares de niños con ST, el 8-57% de los padres tenían antecedentes de tics, y los familiares de primer grado tenían un riesgo 10 a 100 veces mayor de desarrollar el trastorno en comparación con individuos de la población general (9)(10). Lo que evidencia la base genética y el componente de riesgo de esta condición.
Los estudios de ligamiento genético fueron de los primeros en investigar la causa genética del ST mediante el estudio de familias con múltiples miembros afectados, con la premisa de una herencia autosómica dominante(11)(12)(13), sin embargo, los estudios no confirmaron ningún locus específico; por el contrario, los hallazgos demostraron que el ST es un trastorno genéticamente complejo. (14)(15)(16)(17)
Estudios más recientes, de análisis de ligamiento no paramétrico y análisis poblacionales mediante GWAS que tienen en cuenta la heterogeneidad de ST y la posible interacción de los factores de riesgo ambientales, han identificado múltiples loci potenciales (18)(19)(20)(21)(22)(23)(24)(25). Sin embargo, los estudios no han coincidido en reportar la misma región genómica, lo que indica que pueden requerirse diferentes enfoques y tamaños de muestra más grandes para identificar los genes de riesgo. Los genes que han sido identificados en algunos estudios, incluyen la peptidasa 2 de la membrana mitocondrial interna (IMMP2L) (26), el gen dela proteína asociada a contactina 2 (CNTNAP2)(27), SLIT y la proteína similar a NTRK 1 (SLITRK1)(28)(21), neuroligina-4 ligada a X (NLGN4X) (29) y el gen de la histidina descarboxilasa (HDC)(17). Para cada uno de estos genes, la ubicación de la proteína a nivel neuronal difiere: IMMP2L se localiza en la membrana interna mitocondrial, CNTNAP2 se localiza en las regiones yuxtaparanodales en el nodo de Ranvier de axones mielinizados; SLIT, la proteína similar a NTRK 1 y la neuroligina-4 se encuentran en la membrana de neuronas postsinápticas y la histidina descarboxilasa se localiza en el retículo endoplásmico(30).
A la fecha, una mutación puntual (W317X) en el exón 9 del gen HDC asociada a la entidad clínica se ha documentado(17), dicho gen codifica para la enzima encargada de catalizar la síntesis de histamina a partir de histidina. La histamina ha sido implicada en procesos del sistema nervioso central que van desde la modulación de la excitación y la cognición hasta el comportamiento, pero sus funciones biológicas exactas aún no están claras. Aunque esta mutación no ha sido descrita en otros individuos afectados o no fuera de esta familia. Su descubrimiento, agregó la hipótesis de la neurotransmisión histaminérgica como posible etiología del ST y motivó al desarrollo de un ensayo clínico con un antagonista del receptor H3, AZD5213, en adolescentes con ST (ClinicalTrials.gov ID: NCT01904773). El apoyo subsecuente para una "hipótesis de histamina" provino de un estudio europeo de asociación basado en análisis familiar y modelos animales.(31)(32)(33)
Evidencia molecular adicional, resultó del estudio de una familia con un caso de ST en quien se documentó una inversión de novo en el cromosoma 13, con punto de corte en 13q31, a 350 kb del gen SLITRK1. Este mismo grupo examinó 174 pacientes con ST no relacionados y encontró una mutación en el marco de lectura del mismo gen, resultando en una proteína truncada en un individuo con ST y un paciente con tricotilomanía(34). Varios estudios posteriores han examinado la asociación de SLITRK1 con ST mediante la resecuenciación de los probandos, pero no se han documentado mutaciones adicionales en el gen(35)(36)(37)(38)(39); dada la limitante de muestra reducida de estudio, los resultados carecen del poder suficiente para confirmar o refutar de manera concluyente una asociación.
Poco se conoce sobre la función de SLITRK1; sin embargo, lo que se ha descrito indica que SLITRK1 se expresa durante el desarrollo axonal, así como, también se expresa en las neuronas colinérgicas del cuerpo estriado a lo largo de la vida(40). Lo anterior demostrado mediante estudios in vitro, en los que adicionalmente se ha demostrado que la regresión dendrítica esta en relación con los alelos asociados a ST(41).
Los medicamentos disponibles están relacionados con la formación de sinapsis excitadora durante el desarrollo cerebral, incluyen fármacos antidopaminérgicos que abarcan neurolépticos (haloperidol) como antipsicóticos atípicos (aripiprazol o risperidona) y agonistas adrenérgicos (clonidina) (3). Estos medicamentos actúan con eficacia variable y muchos tienen efectos secundarios como sedación, aumento de peso, discinesia o embotamiento cognitivo (11)(42).
Presentamos el caso de un adolescente con antecedente de dos líneas generacionales afectadas con la misma sintomatología, lo que resalta la importancia de realizar una adecuada historia clínica y la realización de un árbol genealógico completo que permita orientar el posible curso de la condición.
Caso Clínico
Paciente masculino de 14 años quien consulta al servicio de neurología pediátrica por cuadro clínico que inicia a los 8 años consistente en parpadeo involuntario repetitivo, encogimiento de hombros y gesticulaciones, ocasional aclaramiento de la voz. No coprolalia, ni ecopraxias. Los síntomas se exacerbaban cuando se encuentra fatigado o estresado, y tienden a ser menos severos cuando realiza actividad física, pero nunca está libre de síntomas. Él puede realizar voluntariamente los tics durante unos minutos. Presentaba como conductas asociadas conducta obsesiva por el lavado de manos desde los 7 años.
Al revisar sus antecedentes, es el producto del primer embarazo de padres jóvenes no consanguíneos, embarazo sin complicaciones, nació por cesárea por pre eclampsia materna, sin complicaciones posnatales; hitos del desarrollo adecuados por rango de edad; desempeño escolar adecuado. Al realizar el árbol genealógico (Figura 1) se evidenció historia familiar del padre, tíos paternos y primo paterno con cuadro clínico de tics fenotípicamente variables que incluían tics oculares, tics de hombro y tics múltiples; la mayoría de ellos actualmente son adultos sin compromiso funcional laboral ni educativo.
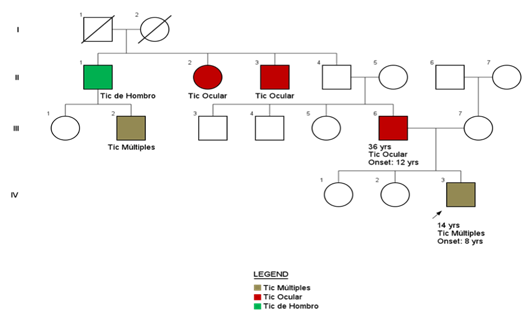
El examen en el momento de su visita reveló un adolescente ansioso con procesos de pensamiento y funcionamiento mental superior intactos. La evaluación del sistema nervioso reveló un parpadeo constante y encogimiento de hombros ocasional. La exploración física no reveló otras anomalías. Durante la consulta se evidenciaron múltiples tics motores en el padre.
Las investigaciones de laboratorio de cuadro hemático, T3L, T4L y TSH, PCR fueron normales, y ASTOS negativos. Dado que los síntomas del paciente no interfirieron significativamente con su desempeño social y académico, se eliminó la medicación como una opción de tratamiento.
Discusión
El síndrome de Tourette (ST) se caracteriza por una reacción repentina e involuntaria de movimientos repetitivos, no rítmicos tics) como parpadear, hacer muecas, sacudir la cabeza o encogerse de hombros. La etiología definitiva de la ST es desconocida, sin embargo, existe evidencia que sugiere la compleja interacción de factores sociales, ambientales y genéticos.
La variabilidad fenotípica y los mecanismos biológicos subyacentes al ST, junto con la necesidad de tratamientos con mayor eficacia, han sido un impulso para comprender la base genética de la enfermedad. El ST no se puede definir simplemente como una enfermedad monogénica o multigénica, sino que es entidad que resulta de los efectos combinados de causas monogénicas, multigénicas y ambientales. Algunos estudios han demostrado la participación de los sistemas dopaminérgicos, glutamatérgicos y serotoninérgicos en la fisiopatología del ST. Más recientemente, la ruta histaminérgica se ha incluido como un nuevo objetivo de investigación y, posiblemente, de tratamiento.
Las opciones de tratamiento para ST varían en efectividad entre los individuos debido a la variabilidad fenotípica y la heterogeneidad genética subyacente. La educación sobre el ST para las personas que interactúan con el paciente es clave para controlar el trastorno. Cuando los síntomas del ST interfieren con el desempeño social, académico u ocupacional se justifica la intervención; dentro de las terapias se contemplan la terapia de comportamiento y varios medicamentos para controlar los tics disruptivos o las comorbilidades como el TDAH, TOC y trastornos del estado de ánimo(43)(44). La terapia conductual que enseña a los pacientes a reconocer los impulsos premonitorios que preceden a los tics, ha demostrado ser efectiva para disminuir la severidad de los tics tanto en presencia como en ausencia de medicamentos; el entrenamiento de respuesta competitiva instruye a los pacientes a completar una acción voluntaria diferente en lugar del tic, mientras que el entrenamiento de relajación se usa para abordar el estrés y la ansiedad, lo que puede tener un impacto en la frecuencia e intensidad de la aparición de tics (43).
El paciente descrito tenía un historial de tics motores complejos de inicio en la infancia asociado a síntomas obsesivos. Este patrón de síntomas cumple con los criterios de diagnóstico para ST (45)(46), una condición que es más frecuente en hombres e involucra principalmente la cara más que otras partes del cuerpo. Su historia familia sugiere un patrón de herencia autosómico dominante con compromiso de 3 generaciones, lo que soporta la hipótesis del componente genético hereditario y los riesgos de recurrencia entre familiares, reportados previamente(9)(10).
El inicio de tratamiento, conductual o farmacológico, se basa en el grado en qué los tics u otros síntomas están interfiriendo en el desarrollo y la educación del niño, o en la funcionalidad laboral y social del adulto. Se debe explicar al paciente y su familia que no hay cura para los tics y todo tratamiento es estrictamente sintomático. En la mayoría de los casos, la medicación es innecesaria. Para el caso reportado, la educación sobre la enfermedad y terapia de comportamiento fueron consideradas como intervenciones de manejo.
Conclusión
A pesar de suponer de una hipótesis genética subyacente al ST por la segregación aparente en las familias y la concordancia en gemelos monocigóticos, actualmente no está indicado realizar estudio genético en individuos con ST dada la posible contribución poligénica de esta entidad. Por tanto, como referente de terapéutica y pronóstico podrían extrapolarse la evolución clínica y las terapias usadas en otros familiares afectados.