









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El Síndrome de Tolosa-Hunt (STH) es una entidad idiopática consistente en una oftalmoplejía dolorosa caracterizada por dolor ocular y por afectación clásica de la primera y segunda rama del trigémino y de uno o más nervios oculomotores (II, IV y V), causada por una inflamación granulomatosa inespecífica del seno cavernoso, de la fisura orbital superior o de la órbita (1)(2). Se caracteriza por ser una condición poco frecuente (entre 1 y 2 casos por cada millón de personas), con reportes aislados a nivel mundial y con una incidencia no establecida (2)(3). En términos de nervios craneales, 70% de los casos presenta compromiso múltiple, principalmente del tercero (79%), sexto (45%), cuarto (32%) y quinto (25%) nervio (3)(4).
Si bien, dicha condición cuenta con una definición y criterios establecidos, actualmente no existe información clara en términos de etiología, asociación con otras patologías, tratamiento, seguimiento a mediano y largo plazo y reincidencia o recaídas (3). Considerando lo anterior, se reporta el caso de una paciente con diagnóstico de Síndrome de Tolosa-Hunt asociado a parálisis facial ipsilateral durante el desarrollo subagudo del cuadro, que contribuye a la comprensión y actualización de la información hasta ahora descrita.
PRESENTACIÓN DEL CASO CLÍNICO
Mujer de 19 años de edad, quien ingresó a una institución de salud de cuarto nivel de complejidad en Santiago de Cali, Colombia, por cuadro de cinco semanas de evolución. Inició con dolor intenso tipo punzada en globo ocular y hemicara derecha, con posterior estrabismo concéntrico derecho y diplopía durante 15 días, seguida de ptosis palpebral derecha. Previo al ingreso, desarrolló parálisis facial periférica derecha y dificultad ipsilateral para masticar, con intensificación del dolor. La paciente negó asociación con disfagia, cambios del tono de voz, tos, vómito o alteraciones de la marcha, sin síntomas en torso o extremidades, ni compromiso de esfínteres; manifestó como único antecedente un consumo frecuente de cannabis y derivados de la cocaína.
Al ingreso presentó midriasis derecha y compromiso de nervios craneales derechos por plejía del III y IV nervio, paresia del VI nervio, hipoestesia-alodinia de las tres ramas sensitivas del V nervio y plejía del músculo masetero derecho, además de parálisis facial periférica derecha (Figura 1). En campimetría y agudeza visual por confrontación no se observaron alteraciones.
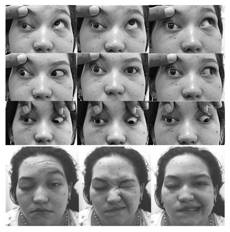
Figura 1 Nueve posiciones diagnósticas de la mirada de la paciente al ingreso. Se observa limitación para aducción, abducción, elevación y depresión del ojo derecho, acompañada de parálisis facial ipsilateral.
Tras valoración médica inicial se realizó resonancia magnética nuclear (RMN) de cerebro con hallazgo de “lesión de apariencia infiltrativa de 2,5 cm x 1,2 cm, hipointensa en T2, con realce homogéneo tras el contraste, que compromete el seno cavernoso derecho y ápex orbitario; la alteración se extiende por el foramen oval a la tercera división del nervio trigémino y afecta posteriormente el borde libre del tentorio y las paquimeninges de la fosa craneal media” (Figura 2). Como parte de las pruebas diagnósticas, a la paciente se le realizó un perfil reumatológico que dio como resultado anticuerpos antinucleares 1/320 diluciones, con patrón nuclear granular fino; el resto del perfil reumatológico fue negativo. El valor de hemoglobina glicosilada fue normal. Se le practicó punción lumbar con presión de apertura de 16 cm H2O: el examen citoquímico mostró una concentración de proteínas de 40 mg/dl, de lactato deshidrogenasa (LDH) menor a 10 unidades/l, de glucosa de 55 mg/dl y un conteo de hematíes de 3/mm3, leucocitos polimorfonucleares de 0/mm3 y linfocitos de 9/mm3. El análisis de líquido cefalorraquídeo no demostró linfocitos atípicos. No se realizó estudio de citometría de flujo (cytospin) para descartar linfoma. Las pruebas ELISA de 4ª generación para VIH y prueba treponémica en sangre fueron negativas, así como la tinta china y el cultivo para bacterias, hongos y tuberculosis. No hubo hallazgos sugestivos de un proceso infeccioso y la linfocitosis se reportó como único hallazgo relevante.
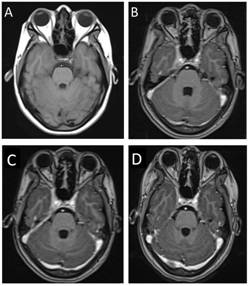
Figura 2 (A) Corte axial a nivel del seno cavernoso de RMN en secuencia T1 simple. (B) Corte axial de RMN en secuencia T1 con Gadolinio, donde se aprecia lesión isointensa en seno cavernoso derecho con realce homogéneo al contraste y compresión del lumen de la arteria carótida. (C) RMN comparativa en secuencia T1 con Gadolinio al ingreso. (D) RMN comparativa en secuencia T1 con Gadolinio a los 8 días de iniciar terapia farmacológica. No se obtuvieron cortes coronales en las secuencias de RMN mencionadas.
Tras haber descartado compromiso sistémico de enfermedad granulomatosa con calcio sérico, tomografía axial computarizada de tórax y biopsia de conjuntiva, se inició manejo con metilprednisolona (1 gr/día por 7 días) y prednisolona (1mg/kg/día) de mantenimiento.
La paciente presentó disminución progresiva del dolor en las primeras 24 horas, con reducción de la lesión del seno cavernoso observada en resonancia nuclear magnética de control a los 8 días de haber iniciado terapia (Figura 2). Se reportó, además, realce del nervio facial ipsilateral en su porción canalicular a nivel de la primera rodilla y del nervio petroso superficial mayor. La paciente mostró mejoría progresiva de los movimientos extraoculares, constricción pupilar y apertura ocular desde la primera semana hasta su egreso, manteniéndose la paresia del músculo recto medial derecho y la parálisis facial ipsilateral (Figura 3).

COMENTARIOS FINALES SOBRE LOS ASPECTOS RELEVANTES DEL CASO
Al considerar la evidencia y la literatura actual en torno al tema, se presenta el reporte de un caso que cumple con los criterios clínicos y radiológicos del STH, con parálisis facial ipsilateral; esta última atribuida probablemente a la extensión del proceso inflamatorio a nivel del segmento distal canalicular y laberíntico del VII par craneal (2). En la mayoría de los estudios no se reporta un compromiso de nervios extracavernosos y compromiso simpático (5)(6)(7)(8). El caso descrito presenta dicha afectación, además del compromiso de las ramas maxilar y mandibular del nervio trigémino, considerado también como una manifestación poco frecuente.
A pesar de contar con más de 60 años de conocimiento, el STH continúa siendo una entidad de etiología desconocida que requiere ser diferenciada de enfermedades de carácter infeccioso, inmuno-reumatológico, neoplásico, traumático, endocrino, iatrogénico, congénito e idiopático, entre las que se destacan los meningiomas, los linfomas, la sarcoidosis y las enfermedades granulomatosas, entre otras (1)(9). El caso resulta ser un reto al considerar diagnósticos diferenciales de oftalmoplejía dolorosa además de condiciones como la parálisis de Bell (7). La paciente presentó dolor orbitario unilateral persistente durante semanas previas al tratamiento, parálisis de los pares craneales descritos, granulomas, oftalmoparesia, dolor y parálisis resuelta en 72 horas posteriores al tratamiento corticoideo; dichos síntomas, descartando otras causas a través de pruebas diagnósticas, son suficientes para la sospecha del STH de acuerdo a los criterios ICHD-II de 1998, revisados en 2004 (3)(6). Sumado a lo anterior, está el consumo previo de sustancias psicoactivas de la paciente, considerado como un antecedente que predispone a la ocurrencia del evento (10).
En términos de ayudas diagnósticas, la RNM resultó ser el examen de elección con una sensibilidad de 92% frente al diagnóstico descrito (3). A través de ésta se busca observar un engrosamiento convexo del seno cavernoso sintomático, isointenso en comparación con la sustancia gris en imágenes TR/TE cortas e hipo-isointenso en imágenes TR/TE largas (3)(9). Es así como, la RMN permitió el diagnóstico y seguimiento de la paciente, lo que apoya la sugerencia de su inclusión como elemento indispensable para un adecuado diagnóstico diferencial respecto a otras patologías como el pseudotumor orbitario caracterizado por engrosamiento de los tendones y músculos oculares, a diferencia del STH (3)(11)(12). Asimismo, junto al descrito por Kang (7), este caso logra demostrar cambios en la intensidad por resonancia magnética del nervio facial afectado, apoyando la hipótesis propuesta de un mecanismo de inflamación focal y distante (6).
En cuanto al tratamiento médico, en 1966 Smith y Taxdall demostraron la dramática respuesta del paciente con STH al manejo con esteroides dentro de las primeras 24 horas (13). Si bien el síntoma dura en promedio 8 semanas, el tiempo se reduce a menos de 72 horas con el uso de esteroides (3)(4). A pesar de no existir consenso en la dosis y vía de administración, la literatura recomienda 1 mg/kg día de prednisona oral y 500-1000 mg/día de metilprednisolona endovenosa. Respecto a la paresia del músculo recto medial derecho y a la parálisis facial ipsilateral de la paciente, éstas puede tardar en desaparecer semanas o meses a pesar del inicio temprano de la terapia corticoide, de acuerdo a la literatura (3). Se recomienda explorar la duración del tratamiento en futuros estudios, al encontrar en la práctica diferentes tiempos de administración. La mejoría clínica es relativamente rápida al iniciar el tratamiento, sin embargo, se sugiere monitorear periódicamente para evaluar la desaparición del tejido en la RNM, puesto que las recurrencias a largo plazo se presentan en aproximadamente la mitad de los pacientes (3)(9).
A manera de conclusión, el Síndrome de Tolosa Hunt es una enfermedad inflamatoria idiopática rara que exige un abordaje diagnóstico complejo, con un amplio espectro de manifestaciones clínicas y paraclínicas compartidas con enfermedades de mayor prevalencia, siendo la parálisis facial un hallazgo poco frecuente. El avance en estudios imagenológicos aporta nuevos hallazgos que pueden contribuir a la caracterización de la etiología, al no contar con prueba diagnóstica definitiva. El tratamiento con esteroides continúa siendo efectivo, con mejora del seno cavernoso y del nervio facial en RNM.