









 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkIntroducción
A partir de la década de los cuarenta del siglo pasado se comenzaron a usar extensamente los pesticidas 4,4’- DDT, Aldrin y Dieldrin, para el control de enfermedades transmitidas por vectores, como la malaria y la enfermedad de Chagas (Aché y Matos, 2001; Aché et al., 2008; Feliciangeli, 2009). Su uso se realizó de forma indiscriminada rociando grandes cantidades de estos compuestos en el interior y exterior de las viviendas, de las cuales aproximadamente el 50% fue dispersado en el medio acuoso, quedando sorbido en sedimentos y suelos. Aunque se dejaron de emplear hace ya varias décadas, por sus características químicas, son resistentes a la degradación y se han convertido en contaminantes universales de aguas, suelos y alimentos (Chen et al., 2007). Además, son lipofílicos, por lo que se acumulan en tejidos biológicos y como consecuencia se magnifica su concentración en individuos que están arriba en la cadena alimenticia (Moreno et al., 2006).
Este comportamiento ha ocasionado que estos pesticidas organoclorados sean clasificados como compuestos orgánicos persistentes (COP) quedando el Aldrin y Dieldrin incluidos en el Anexo A y el 4,4´- DDT en el Anexo B del Convenio de Estocolmo, el cual prohíbe o limita el uso de estos compuestos (COP, 2009). En la actualidad, han sido identificados en muestras de: leche materna, calostro, agua de consumo humano, sangre materna, sangre de neonato, hortalizas, plantas de arroz, pastos, suelos, peces, margarinas, mantequillas, yogures y aceites vegetales distribuidos comercialmente (Alvarado y Yépez, 1998; Urdaneta et al., 2004; Gil, 2006; González et al, 2007; Medina, 2008; Gasull et al, 2011).
La ingeniería de yacimiento se enfoca en adquirir un mejor conocimiento de las características del mismo, para que el ingeniero sea capaz de estimar reservas, predecir el comportamiento del yacimiento con la finalidad de desarrollar modelos de producción.
Su contenido en el ambiente está en el orden de mg/Kg por lo cual, para poder realizar estudios de estos compuestos, es importante cuantificarlos eficientemente y con una adecuada sensibilidad, para ello, es importante diseñar métodos de cuantificación de probada efectividad. En años recientes se han aplicado diversas técnicas analíticas de concentración, extracción y análisis de estos pesticidas en las aguas, tales como CG - EM y HPLC.
El HPLC otorga la ventaja del análisis directo de la fases acuosa, sin embargo, al estar en tan pequeñas concentraciones se requiere el paso previo de concentración. La aplicación de la extracción líquidolíquido en agua, sangre y leche ha sido ampliamente aceptada como método estándar de análisis de pesticidas, ya que es simple, provee un alto grado de limpieza en la muestra y se obtiene una extracción selectiva fácilmente si se elige el solvente adecuado (Cobzac y Gocan, 2011; Xu et al, 2013). Sin embargo, no existe un método único para extraer de soluciones acuosas y cuantificar simultáneamente los pesticidas 4,4’-DDT, Aldrin y Dieldrin por medio de cromatografía líquida de alta resolución con detector de UV-visible (HPLC-UV-visible). Es por ello que en esta investigación el objetivo principal es optimizar la determinación de estos pesticidas en agua por HPLC con detector de UV - visible, empleando como fase móvil acetonitrilo y agua y un método de extracción líquido-líquido con cloroformo, como solvente.
Materiales y métodos
Reactivos
Los patrones sólidos de pesticidas de 4,4’-DDT, Aldrin y Dieldrin fueron obtenidos de Riedel-De Haën AG al ≥ 99% de pureza, el acetonitrilo usado provino de AnalaR Normapur al 99,5% de pureza. , el cloroformo usado se obtuvo de AnalaR Normapur al 99,5% de pureza. Adicionalmente, se emplearon KCl y N2SO4 sólidos al 100% de pureza, obtenidos de JT Baker.
Todos los solventes empleados fueron filtrados a través de un filtro de 0,45 m de membrana de Politetrafluoroetileno (PTFE). El agua empleada fue filtrada con un filtro de acetato de celulosa de 0,45 μm.
Equipos
El sistema de HPLC empleado fue un Agilent 1200 Infinity Series acoplado a un detector de arreglo de diodos (DAD en sus siglas en inglés) Agilent 1200 Infinity Series, con una columna de fase reversa Agilent Eclipse XDB-C18 de 4,6 mm de diámetro interno, 150 mm de longitud y 5 μm de diámetro de partícula y un sistema de inyección manual con loop de inyección de 50 μL. Se trabajó con un flujo de fase móvil de 1 mL/min.
Extracción
10 mL de una mezcla agua: acetonitrilo (50:50 v/v) en la que se sembraron pesticidas, a partir de una madre de 6500 mg/Kg de todos los pesticidas, se pesó y agregó en un embudo de separación de 50 mL, para facilitar la extracción se adicionaron 0,05 g de KCl. La extracción líquido-líquido se realizó con tres extracciones sucesivas usando 5 mL de cloroformo en cada una. El cloroformo fue recolectado en un balón de 50 mL, al que se le agregó 0,5 g de Na2SO4 anhidro para secar la muestra, la solución se transvasó a un balón y se concentró la fracción orgánica empleando un rotaevaporador. Una vez evaporado el solvente, se reconstituyó el extracto orgánico con 2 mL de acetonitrilo, de esta última solución se inyectaron 50 μL en el HPLC-UV-visible.
Soluciones de la curva de calibración
Para realizar la curva de calibración se prepararon soluciones madres de cada pesticida de 6500 mg/Kg en acetonitrilo. Luego se prepararon multipatrones por dilución también en acetonitrilo a las siguientes concentraciones aproximadas para cada pesticida: 0,15; 0,5; 1; 1,5; 3; 5; 10, 15, 35 y 60 mg/Kg. Cada una de estas soluciones se inyectó por triplicado en el HPLCUV- visible generándose las áreas, las cuales se graficaron versus la concentración de cada pesticida. Con la línea recta generada de cada gráfica se calculó la ecuación de la recta, esta ecuación se empleó para calcular la concentración del resultado de cada extracción, de la muestra sembrada, a partir del área de los cromatogramas.
Porcentaje de recobro del método
Para validar el método se preparó una solución de los tres pesticidas en una mezcla acetonitrilo: agua (50:50v/v), para simular una matriz natural que puede ser capaz de disolver gran cantidad de pesticidas, lo que no ocurre en agua pura. Se evaluaron dos niveles de concentración 2 y 20 mg/Kg. En estas soluciones se aplicó el proceso de extracción líquido-líquido y por último fueron analizadas por HPLC-UV-visible.
Límite de detección y límite de cuantificación
Para calcular el límite de detección (LOD) y límite de cuantificación (LOQ) para cada pesticida se analizó a través del HPLC-UV-visible el blanco en seis ocasiones y se calculó: el promedio y la desviación estándar de las señales del blanco para cada pesticida (Sbl y sbl respectivamente), los resultados de estos cálculos se usaron para determinar la mínima señal analítica distinguible (Sm) y la mínima señal analítica cuantificable (SQ) para cada uno de los pesticidas usando las ecuaciones 1 y 2:
Sm = Sbl + 3 sbl (Ecuación 1)
SQ = Sbl + 10 sbl (Ecuación 2)
A continuación, se sustituyeron los valores de Sm y SQ en cada una de las ecuaciones de la recta obtenidas a partir de las curvas de calibración de cada pesticida, pero colocando como intercepto el promedio de la señal del blanco (Sbl), para calcular LOD o la más baja concentración del analito en la muestra que puede ser detectada más no cuantificada y el LOQ, o la concentración mínima a la cual se pueden efectuar mediciones cuantitativas. Con esta metodología se puede calcular estadísticamente que el nivel de confianza en la detección será de un 95 % en la mayoría de los casos (Miller y Miller, 2001; Skoog et al, 2008; Christian, 2009).
Resultados y discusión
Desarrollo del método
El estudio preliminar involucró la selección de la fase móvil. Se probaron tres (3) opciones para esta fase: acetonitrilo: agua 50:50, acetonitrilo: agua 80:20 y acetonitrilo: agua 85:15. Para determinar el tiempo de retención se inyectó un pesticida por vez para cada fase móvil de forma isocrática. Posteriormente, se escogió la longitud de onda de mayor absorbancia para cada pesticida realizando un barrido de longitudes de onda del rango del espectro UV-visible. Para cada longitud de onda seleccionada se determinaron los parámetros que mostraron mayor sensibilidad y selectividad, estos se pueden observar en la tabla 1. En cuanto al volumen de inyección se decidió usar un loop de inyección de 50 μL porque a concentraciones menores a 1 mg/Kg el Aldrin no era detectado.
Tabla 1 Tiempos de retención, longitud de onda y parámetros cromatográficos de mayor sensibilidad para cada pesticida.
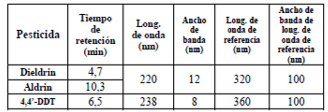
La primera opción de fase móvil fue la mezcla acetonitrilo: agua (50:50), la cual fue descartada porque los tiempos de retención de los pesticidas no se mantenían constantes al realizar repeticiones y se presentaba un gran ensanchamiento de los picos cromatográficos. La segunda opción de fase móvil fue la mezcla acetonitrilo: agua (80:20), en la cual si se observaba constancia en los tiempos de retención al realizar repeticiones. Sin embargo, el Aldrin, el cual es el último compuesto en eludir en el cromatograma, presentaba un gran ensanchamiento, de más de 1 minuto.
Por lo tanto, se decidió aumentar el porcentaje de acetonitrilo en la fase móvil llegando a la relación acetonitrilo: agua (85:15), con esta mezcla se logró que los tiempos de retención se mantuvieran constantes al realizar repeticiones y que no se presentaran ensanchamiento de los picos cromatográficos, en la figura 1 se puede observar un cromatograma, en el cual se aprecia la buena selectividad del método empleado.

Método de validación
Cada solución empleada para realizar la curva de calibración se inyectó por triplicado en el HPLC-UVvisible, se promediaron los tres resultados y se calculó la desviación estándar entre las áreas obtenidas del cromatograma; dichas áreas se graficaron versus la concentración de las soluciones preparadas a partir de patrones. En las figuras 2, 3 y 4 se pueden observar las curvas de calibración generadas por estos datos, con la ecuación de la recta y el valor de R2. En estas gráficas de áreas de los cromatogramas versus concentración se observa una respuesta lineal en el rango de 0,27 - 62mg/Kg para Diledrín, con un R2 de 0,996, de 0,14 - 42 mg/Kg para 4,4'-DDT con un R2 de 0,999 y un rango de 0,34 - 81 mg/Kg para Aldrin y un R2 de 0,999.
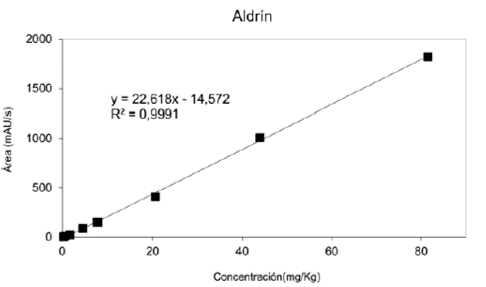
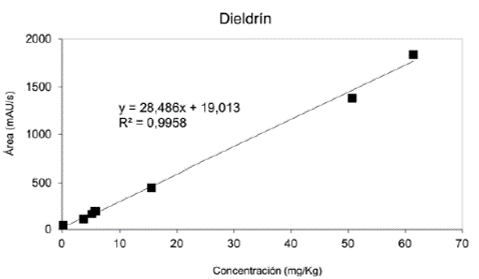
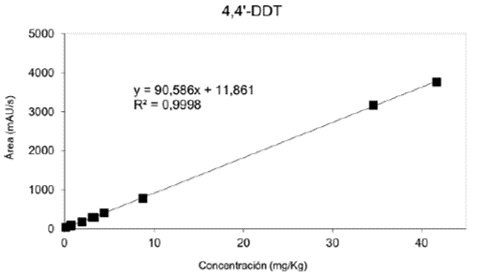
Recobro
Para evaluar la exactitud del método empleado se calculó el porcentaje de recobro de los tres pesticidas estudiados, sembrados en una matriz de acetonitrilo: agua (50:50). Esto se realizó para dos concentraciones, 2 y 20 mg/Kg, y sus réplicas. El porcentaje de recobro se calculó por la relación entre el área obtenida de la extracción y análisis del pesticida sembrado y el área teórica de la solución preparada con un patrón. El porcentaje de desviación estándar calculado a partir de las réplicas permite estimar la precisión del método. Los resultados para la concentración de 20 mg/Kg se presentan en la tabla 2. El método empleado tiene un porcentaje de recobro en el rango de 91 - 104 %, para los tres pesticidas en los dos niveles de concentración estudiados (2 y 20 mg/Kg) y con una desviación estándar relativa en el rango de 0,8 - 8%.
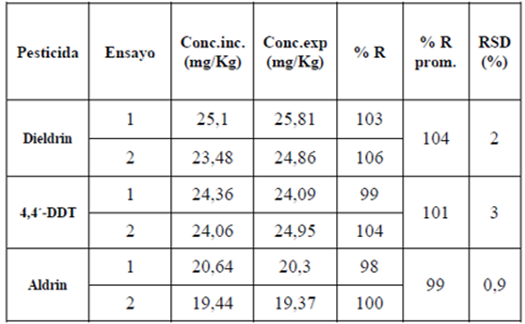
Límite de detección y de cuantificación
Los límites de detección y cuantificación obtenidos de los tres pesticidas se muestran en la tabla 3. Los valores obtenidos son bajos lo cual permite la determinación y cuantificación de estos compuestos por el método descrito, en aguas a bajos niveles de concentración.
Tabla 3 Límite de detección (LOD) y límite de cuantificación (LOQ) de los pesticidas analizados.
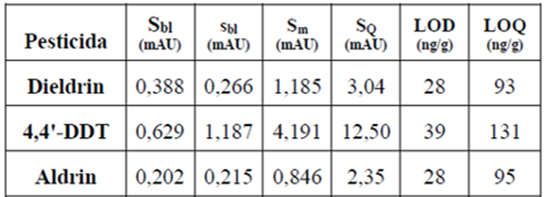
Sbl: concentración promedio para el blanco; sbl: desviación estándar promedio para el blanco; Sm: mínima señal analítica detectable; SQ: mínima señal analítica cuantificable
En vista de que los valores máximos permitidos de estos compuestos en aguas están en el orden de 0,1 - 2 g/L (Ballesteros, 2007; Lehotay y Mastovska, 2005), es necesario la concentración con solventes como el cloroformo, de volúmenes de muestra de agua en el orden de 100 mL, para con los LOD y LOQ obtenidos en esta metodología identificar estos pesticidas y luego cuantificarlos.
Conclusiones
Un método simple, exacto y preciso de HPLC-UVvisible fue desarrollado y validado para la determinación de tres pesticidas organoclorados(4,4’- DDT, Aldrin y Dieldrin) en aguas. El método es óptimo en un amplio rango lineal (0,27 - 62 mg/Kg para Diledrin, 0,14 - 42 mg/Kg para 4,4'-DDT y 0,34 - 81 mg/Kg para Aldrin) con porcentaje de recobro de 91 - 104 %. Los bajos valores de LOD y LOQ permiten la determinación y cuantificación de estos pesticidas en aguas naturales a bajas concentraciones.